PHYSIOPATHOLOGIE DES MALADIES INFLAMMATOIRES CHRONIQUES ET BIOTHÉRAPIES ASSOCIÉES
Acronyme PACIDbio
Coordonnateurs: POUPOT Remy et AUSSEIL Jérôme
Objectifs scientifiques:
Les maladies inflammatoires et neuro-inflammatoires chroniques sont caractérisées par un système immunitaire dérégulé. Parmi toutes les cellules immunitaires, les cellules myéloïdes apparaissent comme des acteurs importants dans ces maladies inflammatoires en raison de leur capacité à favoriser l’inflammation et le stress oxydatif, en plus de leur capacité à détecter et à atténuer les états pathogènes. Notre équipe s’intéresse à la microglie cérébrale et aux cellules monocytes / macrophages dans les tissus qui sont également confrontés à des inflammations locales chroniques telles que: l’os, les articulations et les reins. Dans ce contexte, la microglie et les macrophages établissent des interactions avec les cellules tissulaires voisines (en libérant des médiateurs ou non) et influencent leurs activités non seulement dans des conditions pathologiques, mais également dans des conditions physiologiques, en particulier pendant le développement, les processus tissulaires homéostatiques, la réparation tissulaire et l’immunité. Ayant un rôle majeur dans l’initiation et la propagation de l’inflammation chronique, la microglie ainsi que les monocytes / macrophages deviennent des cibles thérapeutiques pertinentes.
Nos recherches se concentrent essentiellement sur la physiopathologie inflammatoires et neuro-inflammatoires, et sur la conception, le développement de nouvelles biothérapies et leur mise en œuvre aux étapes précliniques et cliniques.
Axe 4 : Remodelage osseux, arthrite et thérapeutique
 Les cellules myéloïdes (monocytes, cellules dendritiques, macrophages et ostéoclastes) jouent un rôle central dans la physiopathologie des maladies inflammatoires ostéoarticulaires telles que la Polyarthrite rhumatoïde et le Rhumatisme psoriasique. Ces cellules sont notamment impliquées dans l’inflammation articulaire, les dommages structuraux osseux et la perte osseuse inflammatoire. Notre objectif est de mieux caractériser l’implication des cellules myéloïdes dans ces processus, ainsi que leur modulation par les traitements actuels ou en développement pour les maladies inflammatoires ostéoarticulaires. Nous interagissons de manière privilégiée avec le Centre de Rhumatologie du CHU de Toulouse et avec le réseau national des Centres Hospitalo-Universitaires de Rhumatologie. Ces liens nous permettent de développer une recherche « translationnelle » au travers de l’étude cohortes de patients, tant à l’échelle locale (Maladies inflammatoires osseuses et articulaires – BIOTOUL), qu’à l’échelle nationale (Polyarthrite rhumatoïde – ESPOIR, Rhumatisme psoriasique – APACHE, Spondyloarthrites – DESIR, Mastocytose systémique – CEREMAST).
Les cellules myéloïdes (monocytes, cellules dendritiques, macrophages et ostéoclastes) jouent un rôle central dans la physiopathologie des maladies inflammatoires ostéoarticulaires telles que la Polyarthrite rhumatoïde et le Rhumatisme psoriasique. Ces cellules sont notamment impliquées dans l’inflammation articulaire, les dommages structuraux osseux et la perte osseuse inflammatoire. Notre objectif est de mieux caractériser l’implication des cellules myéloïdes dans ces processus, ainsi que leur modulation par les traitements actuels ou en développement pour les maladies inflammatoires ostéoarticulaires. Nous interagissons de manière privilégiée avec le Centre de Rhumatologie du CHU de Toulouse et avec le réseau national des Centres Hospitalo-Universitaires de Rhumatologie. Ces liens nous permettent de développer une recherche « translationnelle » au travers de l’étude cohortes de patients, tant à l’échelle locale (Maladies inflammatoires osseuses et articulaires – BIOTOUL), qu’à l’échelle nationale (Polyarthrite rhumatoïde – ESPOIR, Rhumatisme psoriasique – APACHE, Spondyloarthrites – DESIR, Mastocytose systémique – CEREMAST).
Nos axes actuels de recherche sont :
- Les mécanismes du Reverse signalling induit par les agents anti-TNF et son implication dans la réponse clinique aux anti-TNF.
- Le rôle de l’Hepcidine dans l’inflammation ostéoarticulaire et le remodelage osseux, durant l’arthrite.
- L’impact des traitements anti-rhumatismaux (DMARDs) sur la polarisation du macrophage et l’ostéoclastogenèse dans les maladies inflammatoires ostéoarticulaires.
Axe 5 : Le syndrome de Prader-Willi, neurodéveloppement et épigénétique
 Le syndrome de Prader Willi (SPW) est une maladie neuro-développementale rare avec des troubles alimentaires sévères ainsi que des troubles du comportement. Nous nous intéressons à la physiopathologie du SPW dans le cadre du Centre National de Référence du SPW, au CHU de Toulouse, coordonné par le Prof. M. Tauber. Nous utilisons des approches complémentaires avec des études de recherche clinique et des modèles cellulaires incluant des iPSC (cellules souches pluripotentes induites) de patients atteints de SPW. Plusieurs études suggèrent une implication d’un cluster de petits ARN nucléolaires, le SNORD116, dans la neuro-inflammation et la neuro-dégénérescence liées au SPW.
Le syndrome de Prader Willi (SPW) est une maladie neuro-développementale rare avec des troubles alimentaires sévères ainsi que des troubles du comportement. Nous nous intéressons à la physiopathologie du SPW dans le cadre du Centre National de Référence du SPW, au CHU de Toulouse, coordonné par le Prof. M. Tauber. Nous utilisons des approches complémentaires avec des études de recherche clinique et des modèles cellulaires incluant des iPSC (cellules souches pluripotentes induites) de patients atteints de SPW. Plusieurs études suggèrent une implication d’un cluster de petits ARN nucléolaires, le SNORD116, dans la neuro-inflammation et la neuro-dégénérescence liées au SPW.
Plus précisément, nous étudions le rôle du SNORD116 dans les anomalies neuroendocriniennes et endocriniennes du SPW. En utilisant des neurones hypothalamiques spécifiques dérivés d’iPSC d’un patient qui présente une délétion très limitée du SNOD116 (Am J Hum Genet, 2015), nous avons contribué à la démonstration du rôle de SNORD116 dans le traitement de plusieurs prohormones (J Clin Invest, 2017; Coll. R. Liebel, Columbia University ; États-Unis).
Nos résultats les plus récents démontrent aussi l’impact du SNORD116 et de l’IGFBP7 sur le traitement par hormone de croissance dans le SPW (Genet Med, 2021). D’autres études sont menées afin d’élucider les mécanismes moléculaires impliqués.
Un autre sujet majeur concerne le dysfonctionnement hypothalamique, en particulier lié aux voies de la ghreline et de l’ocytocine (OXT) dans une perspective thérapeutique. Nous avons démontré les effets bénéfiques de l’OXT administré aux nourrissons atteints de SPW (Pediatrics, 2018 et Rev. in Lancet Diabetes Endocrinol, 2021). Les voies de la ghreline et de l’OXT dans le SPW en relation avec l’absence du SNORD116 sont également fortement impliquées dans l’addiction, la prise alimentaire et les troubles du comportement, ceci potentiellement liés à des modifications épigénétiques (Mol Psychiatry, 2020 et Transl Psychiatry, 2020).
Nous menons une étude prospective avec placebo sur l’effet de l’OXT chez le nourrisson. Elle est développée dans le cadre d’un essai européen PEDCRIN (OTBB3 Trial PedCRIN H2020 GA- N ° 731046 4) et soutenue par la startup OT4B. Cette étude sera un support pour clarifier l’impact du SNORD116 sur les processus épigénétiques dans les voies sous-jacentes au circuit dopaminergique de la récompense impliqué dans l’addiction et dans l’interaction sociale dans le SPW (J. Salles, thèse de doctorat).
Un autre sujet aborde le rôle des perturbations de la ghreline et de la résistance hormonale dans le métabolisme osseux. En ce qui concerne la physiopathologie de la scoliose fréquemment observée dans les patients atteints du SPW, les mécanismes moléculaires impliquant la résistance cellulaire sont testés dans des modèles d’ostéoblastes (Biochim. Biophys. Acta, 2020 et Biochem Biophys Rep, 2020). Ceci est en lien avec le Centre de Référence du Métabolisme du Calcium et du Phosphate et ERN-Bone coordonné par le Prof. J. P. Salles.
Axe 6 : AlterAG, nouvelle voie de synthèse de l'endocannabinoïde 2-arachidonoylglycérol et pathologies infectieuses et inflammatoires
 La découverte du système endocannabinoïde (ECS), révélé à partir d’études sur le composé psychotrope le plus puissant de Cannabis sativa, le D-9-tétrahydrocannabinol (THC), a ouvert un nouveau domaine de pharmacologie suggérant son potentiel thérapeutique dans diverses conditions pathologiques. L’ECS est constitué des récepteurs cannabinoïdes, de leurs ligands endogènes (endocannabinoïdes) et des protéines contrôlant leur synthèse et leur dégradation. À ce jour, deux récepteurs cannabinoïdes (CB1 et CB2) ont été identifiés. Ce sont des récepteurs membranaires cellulaires contenant sept domaines transmembranaires appartiennant à la famille des récepteurs couplés aux protéines G. Leur expression est tissu-spécifique puisque CB1 est principalement localisé dans le système nerveux central tandis que CB2 est principalement exprimé dans le système immunitaire. Les deux récepteurs sont activés par des médiateurs lipidiques endogènes qui sont libérés «à la demande» en réponse à divers stimuli physiologiques et pathologiques. Les cannabinoïdes ont des activités pléiotropes telles que des propriétés anti-inflammatoires, immunomodulatrices et neuroprotectrices. Nous avons découvert une nouvelle voie de synthèse du principal endocannabinoïde, le 2-arachidonoylglycérol (2-AG), l’agoniste complet des deux récepteurs. Elle implique l’hydrolyse par une lysophospholipase C appelée GDE3 (gène GDPD2) du lysophosphatidylinositol (LPI), lui-même ligand d’un troisième type de récepteur endocannabinoïde (GPR55). GDE3 est exprimée notamment dans les fibroblastes spléniques de pulpe rouge, les entérocytes de l’intestin grêle, la peau et une sous-population d’astrocytes.Profitant de l’utilisation de souris invalidées pour GDPD2 (GDE3-KO), nous avons montré un rôle de GDE3 dans la défense contre les bactéries circulantes, notamment au niveau de la rate. GDE3 pourrait également contribuer aux processus neuro-inflammatoires en intervenant à deux niveaux: l’intestin grêle, où elle est essentielle au maintien de l’intégrité de la barrière intestinale et de l’équilibre du microbiote, le système nerveux central, où elle assure la production de 2-AG sur des sites spécifiques qui restent à identifié.
La découverte du système endocannabinoïde (ECS), révélé à partir d’études sur le composé psychotrope le plus puissant de Cannabis sativa, le D-9-tétrahydrocannabinol (THC), a ouvert un nouveau domaine de pharmacologie suggérant son potentiel thérapeutique dans diverses conditions pathologiques. L’ECS est constitué des récepteurs cannabinoïdes, de leurs ligands endogènes (endocannabinoïdes) et des protéines contrôlant leur synthèse et leur dégradation. À ce jour, deux récepteurs cannabinoïdes (CB1 et CB2) ont été identifiés. Ce sont des récepteurs membranaires cellulaires contenant sept domaines transmembranaires appartiennant à la famille des récepteurs couplés aux protéines G. Leur expression est tissu-spécifique puisque CB1 est principalement localisé dans le système nerveux central tandis que CB2 est principalement exprimé dans le système immunitaire. Les deux récepteurs sont activés par des médiateurs lipidiques endogènes qui sont libérés «à la demande» en réponse à divers stimuli physiologiques et pathologiques. Les cannabinoïdes ont des activités pléiotropes telles que des propriétés anti-inflammatoires, immunomodulatrices et neuroprotectrices. Nous avons découvert une nouvelle voie de synthèse du principal endocannabinoïde, le 2-arachidonoylglycérol (2-AG), l’agoniste complet des deux récepteurs. Elle implique l’hydrolyse par une lysophospholipase C appelée GDE3 (gène GDPD2) du lysophosphatidylinositol (LPI), lui-même ligand d’un troisième type de récepteur endocannabinoïde (GPR55). GDE3 est exprimée notamment dans les fibroblastes spléniques de pulpe rouge, les entérocytes de l’intestin grêle, la peau et une sous-population d’astrocytes.Profitant de l’utilisation de souris invalidées pour GDPD2 (GDE3-KO), nous avons montré un rôle de GDE3 dans la défense contre les bactéries circulantes, notamment au niveau de la rate. GDE3 pourrait également contribuer aux processus neuro-inflammatoires en intervenant à deux niveaux: l’intestin grêle, où elle est essentielle au maintien de l’intégrité de la barrière intestinale et de l’équilibre du microbiote, le système nerveux central, où elle assure la production de 2-AG sur des sites spécifiques qui restent à identifié.
L’objectif de cet axe est d’étudier le rôle de GDE3 et les mécanismes impliqués dans plusieurs modèles murins de pathologies infectieuses et inflammatoires du tractus gastro-intestinal, du cerveau (Encéphalomyélite auto-immune expérimentale ou EAE, maladie de Parkinson et syndrome de Sanfilippo), de la peau (psoriasis) et du système immunitaire. L’implication de la voie alternative de production de 2-AG, que nous appelons Alter-AG, pourrait ainsi fournir des cibles pharmacologiques intéressantes à explorer dans le cadre de ces pathologies.

AXE 1: Etude physiopathologique du syndrome de Sanfilippo (Mucopolysaccharidose de type III) et approches thérapeutiques par thérapie génique.
 Le syndrome de Sanfilippo est caractérisé par une neuroinflammation sévère associée à une neurodégénérescence provoquée par l’absence d’une exoglycanase lysosomale conduisant à l’interruption de la dégradation de l’héparane sulfate (HS). L’accumulation progressive d’oligosaccharides d’HS (HSO) induit le développement d’un stress oxydatif, la production de cytokines et de chimiokines via une activation dépendante du récepteur TLR4 dans le système nerveux central (SNC) (Trudel, J Neurosci Res 2015) et dans le système ostéoarticulaire. L’activation du TLR4 a plusieurs conséquences dont l’induction d’une signalisation microgliale de STAT3 conduisant à une augmentation de l’expression de l’hepcidine et à une rétention de fer dans le cerveau (Puy, Glia 2018). Nous étudions actuellement les états fonctionnel et moléculaire de la microglie dans ce syndrome ainsi que l’implication de la microglie dans l’initiation et la propagation de l’inflammation conduisant aux processus neurodégénératifs (étude réalisée sur modèle cellulaire BV2 murine, microglie adulte primaire, microglie humaine CHME-5 et microglie primaire provenant de souris Sanfilippo B). Nos récents résultats indiquent que les vésicules extracellulaires (EVs) dérivées de la microglie seraient impliquées dans la propagation des signaux inflammatoires dans le cerveau. Cependant, on en sait peu sur la biogenèse et la régulation de cette production d’EVs. En utilisant des approches biochimiques et protéomiques, nous étudions la production, la composition et le rôle de ces EVs microgliales produites au cours de la neuroinflammation.
Le syndrome de Sanfilippo est caractérisé par une neuroinflammation sévère associée à une neurodégénérescence provoquée par l’absence d’une exoglycanase lysosomale conduisant à l’interruption de la dégradation de l’héparane sulfate (HS). L’accumulation progressive d’oligosaccharides d’HS (HSO) induit le développement d’un stress oxydatif, la production de cytokines et de chimiokines via une activation dépendante du récepteur TLR4 dans le système nerveux central (SNC) (Trudel, J Neurosci Res 2015) et dans le système ostéoarticulaire. L’activation du TLR4 a plusieurs conséquences dont l’induction d’une signalisation microgliale de STAT3 conduisant à une augmentation de l’expression de l’hepcidine et à une rétention de fer dans le cerveau (Puy, Glia 2018). Nous étudions actuellement les états fonctionnel et moléculaire de la microglie dans ce syndrome ainsi que l’implication de la microglie dans l’initiation et la propagation de l’inflammation conduisant aux processus neurodégénératifs (étude réalisée sur modèle cellulaire BV2 murine, microglie adulte primaire, microglie humaine CHME-5 et microglie primaire provenant de souris Sanfilippo B). Nos récents résultats indiquent que les vésicules extracellulaires (EVs) dérivées de la microglie seraient impliquées dans la propagation des signaux inflammatoires dans le cerveau. Cependant, on en sait peu sur la biogenèse et la régulation de cette production d’EVs. En utilisant des approches biochimiques et protéomiques, nous étudions la production, la composition et le rôle de ces EVs microgliales produites au cours de la neuroinflammation.
Il n’y a pour le moment aucune thérapie pour ces pathologies, l’espérance de vie ne dépassant pas la vingtaine pour ces enfants. En collaboration avec des équipes de l’Institut Pasteur et de l’APHP, nous avons participé à un premier essai clinique de phase I / II de thérapie génique intracérébrale sur 4 enfants Sanfilippo B dont les résultats sont très encourageants. Cependant, le bénéfice neurocognitif n’étant que partiel, probablement parce que l’enzyme n’est pas délivrée à la périphérie du cerveau.
Notre équipe relance une préclinique sur modèles animaux. Si les résultats sont probants, un nouvel essai clinique sera programmé. Ce projet est financé par l’association Vaincre les Maladies Lysosomales.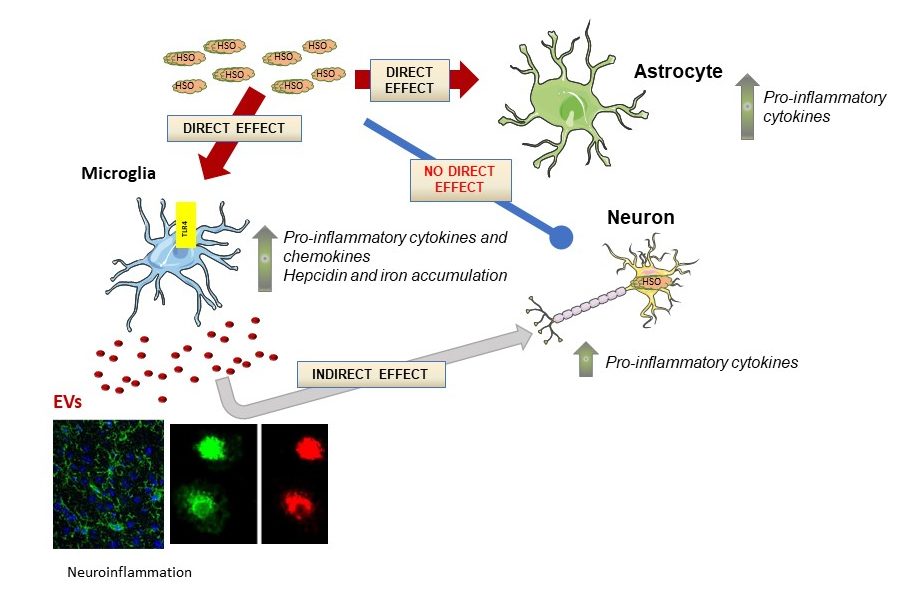
AXE 3: Métabolisme du fer et l’hepcidine dans les maladies inflammatoires chroniques
 Le fer présent dans toutes les cellules de l’organisme, est nécessaire à la vie (transport d’oxygène, réactions de transfert d’électrons ou synthèse d’ADN). Le fer physiologique est principalement utilisé pour produire de l’hème, le groupement prothétique des hémoprotéines telles que l’hémoglobine, la myoglobine et le cytochrome P450. Cependant, l’excès de fer représente une réelle menace pour la cellule car elle est capable de générer des radicaux libres (ou «réactifs oxygénés» (ROS)) lors de la réaction de Fenton (réaction chimique utilisant du fer et de l’eau oxygénée pour produire un radical hydroxyle: H2O2 + Fe2 + « OH- + OH • + Fe3 +). En plus, le fer peut également favoriser l’infection pathogène. Dans l’organisme, l’équilibre du fer est assuré par un équilibre strict entre son absorption par l’intestin et son stockage et recyclage par les macrophages suite à la phagocytose des globules rouges sénescents et à la dégradation de qui de l’hémoglobine et l’hème. L’absorption intestinale et la libération de fer des macrophages sont contrôlées par l’hepcidine, un petit peptide synthétisé et sécrété par le foie. L’expression de l’hepcidine est régulée par les variations du fer dans l’organisme (induites par une surcharge en fer et réprimée par une carence en fer et une érythropoïèse stimulée). De plus, étant une protéine de phase aiguë, l’hepcidine est induite par l’inflammation, ce qui en pathologie contribue à une carence en fer fonctionnelle et à une anémie inflammatoire. L’hepcidine est également exprimée dans de nombreux autres organes, bien qu’à un niveau beaucoup plus bas (rein, cerveau, os, monocytes / macrophages).
Le fer présent dans toutes les cellules de l’organisme, est nécessaire à la vie (transport d’oxygène, réactions de transfert d’électrons ou synthèse d’ADN). Le fer physiologique est principalement utilisé pour produire de l’hème, le groupement prothétique des hémoprotéines telles que l’hémoglobine, la myoglobine et le cytochrome P450. Cependant, l’excès de fer représente une réelle menace pour la cellule car elle est capable de générer des radicaux libres (ou «réactifs oxygénés» (ROS)) lors de la réaction de Fenton (réaction chimique utilisant du fer et de l’eau oxygénée pour produire un radical hydroxyle: H2O2 + Fe2 + « OH- + OH • + Fe3 +). En plus, le fer peut également favoriser l’infection pathogène. Dans l’organisme, l’équilibre du fer est assuré par un équilibre strict entre son absorption par l’intestin et son stockage et recyclage par les macrophages suite à la phagocytose des globules rouges sénescents et à la dégradation de qui de l’hémoglobine et l’hème. L’absorption intestinale et la libération de fer des macrophages sont contrôlées par l’hepcidine, un petit peptide synthétisé et sécrété par le foie. L’expression de l’hepcidine est régulée par les variations du fer dans l’organisme (induites par une surcharge en fer et réprimée par une carence en fer et une érythropoïèse stimulée). De plus, étant une protéine de phase aiguë, l’hepcidine est induite par l’inflammation, ce qui en pathologie contribue à une carence en fer fonctionnelle et à une anémie inflammatoire. L’hepcidine est également exprimée dans de nombreux autres organes, bien qu’à un niveau beaucoup plus bas (rein, cerveau, os, monocytes / macrophages).
L’objectif principal de cet axe 3 est de développer des recherches conduisant à élucider le rôle de l’hepcidine extra-hépatique dans le processus d’infection / inflammation et dans les pathologies confrontées au stress oxydatif dû à l’accumulation incontrôlée de fer inflammatoire, tant chez les modèles murins que chez les patients. Actuellement, nos priorités sont les maladies rénales (anémie inflammatoire, infection des voies urinaires), la neuroinflammation (syndrome de Sanfilippo) et l’os / articulations (maladie rhumatismale).

AXE 2: Physiopathologie de la maladie de Parkinson et nouvelles thérapies anti-inflammatoires dans les maladies neurodégéneratives.

La «microgliose réactive» est maintenant considérée comme l’agent causal de la neuroinflammation qui conduit à la neurodégénérescence dans plusieurs maladies liées au vieillissement (y compris la maladie d’Alzheimer et la maladie de Parkinson, PD). Une partie de nos recherches consiste à identifier les mécanismes et la cinétique conduisant à la «microgliose réactive» dans la PD en utilisant un modèle murin. Nous concevons également de nouvelles approches thérapeutiques pour traiter l’inflammation chronique en ciblant les cellules myéloïdes (monocytes / macrophages, cellules dendritiques, etc.). Nous avons montré que les dendrimères immuno-modulateurs à base phosphorée tels que le dendrimère ABP, sont capables de basculer l’activation pro-inflammatoire des monocytes / macrophages vers un phénotype anti-inflammatoire. Nous avons prouvé l’efficacité thérapeutique de ces molécules dans plusieurs modèles murins de maladies inflammatoires chroniques (polyarthrite rhumatoïde, sclérose en plaques, etc.).
Nous voulons maintenant tester l’efficacité thérapeutique du dendrimère ABP (ou analogues bioactifs) dans des modèles murins de maladies neurodégénératives. Les mécanismes cellulaires et moléculaires sous-tendant l’efficacité thérapeutique des dendrimères, le cas échéant, seront définis. Parallèlement, nous collaborons avec l’équipe de Michel Simon chez INFINITy pour explorer les propriétés anti-inflammatoires des dendrimères phosphorés pour traiter les maladies inflammatoires de la peau, et en particulier le psoriasis.
Autres informations
Notre équipe
Publications
2024 |
Dias, Chloé; Ballout, Nissrine; Morla, Guillaume; Alileche, Katia; Santiago, Christophe; Guerrera, Ida Chiara; Chaubet, Adeline; Ausseil, Jerome; Trudel, Stephanie Extracellular vesicles from microglial cells activated by abnormal heparan sulfate oligosaccharides from Sanfilippo patients impair neuronal dendritic arborization Article de journal Dans: Molecular Medicine, vol. 30, no. 1, p. 197, 2024, ISSN: 1528-3658. @article{nokey,Background In mucopolysaccharidosis type III (MPS III, also known as Sanfilippo syndrome), a pediatric neurodegenerative disorder, accumulation of abnormal glycosaminoglycans (GAGs) induces severe neuroinflammation by triggering the microglial pro-inflammatory cytokines production via a TLR4-dependent pathway. But the extent of the microglia contribution to the MPS III neuropathology remains unclear. Extracellular vesicles (EVs) mediate intercellular communication and are known to participate in the pathogenesis of adult neurodegenerative diseases. However, characterization of the molecular profiles of EVs released by MPS III microglia and their effects on neuronal functions have not been described. Methods Here, we isolated EVs secreted by the microglial cells after treatment with GAGs purified from urines of Sanfilippo patients (sfGAGs-EVs) or from age-matched healthy subjects (nGAGs-EVs) to explore the EVs’ proteins and small RNA profiles using LC–MS/MS and RNA sequencing. We next performed a functional assay by immunofluorescence following nGAGs- or sfGAGs-EVs uptake by WT primary cortical neurons and analyzed their extensions metrics after staining of βIII-tubulin and MAP2 by confocal microscopy. Results Functional enrichment analysis for both proteomics and RNA sequencing data from sfGAGs-EVs revealed a specific content involved in neuroinflammation and neurodevelopment pathways. Treatment of cortical neurons with sfGAGs-EVs induced a disease-associated phenotype demonstrated by a lower total neurite surface area, an impaired somatodendritic compartment, and a higher number of immature dendritic spines. Conclusions This study shows, for the first time, that GAGs from patients with Sanfilippo syndrome can induce microglial secretion of EVs that deliver a specific molecular message to recipient naive neurons, while promoting the neuroinflammation, and depriving neurons of neurodevelopmental factors. This work provides a framework for further studies of biomarkers to evaluate efficiency of emerging therapies. |
Salles, Juliette; Eddiry, Sanaa; Amri, Saber; Galindo, Mélissa; Lacassagne, Emmanuelle; George, Simon; Mialhe, Xavier; Lhuillier, Émeline; Franchitto, Nicolas; Jeanneteau, Freddy; Gennero, Isabelle; Salles, Jean Pierre; Tauber, Maithé Dans: Molecular Psychiatry, no. March, 2024, ISSN: 14765578. @article{Salles2024,Introduction: A microdeletion including the SNORD116 gene (SNORD116 MD) has been shown to drive the Prader-Willi syndrome (PWS) features. PWS is a neurodevelopmental disorder clinically characterized by endocrine impairment, intellectual disability and psychiatric symptoms such as a lack of emotional regulation, impulsivity, and intense temper tantrums with outbursts. In addition, this syndrome is associated with a nutritional trajectory characterized by addiction-like behavior around food in adulthood. PWS is related to the genetic loss of expression of a minimal region that plays a potential role in epigenetic regulation. Nevertheless, the role of the SNORD116 MD in DNA methylation, as well as the impact of the oxytocin (OXT) on it, have never been investigated in human neurons. Methods: We studied the methylation marks in induced pluripotent stem-derived dopaminergic neurons carrying a SNORD116 MD in comparison with those from an age-matched adult healthy control. We also performed identical neuron differentiation in the presence of OXT. We performed a genome-wide DNA methylation analysis from the iPSC-derived dopaminergic neurons by reduced-representation bisulfite sequencing. In addition, we performed RNA sequencing analysis in these iPSC-derived dopaminergic neurons differentiated with or without OXT. Results: The analysis revealed that 153,826 cytosines were differentially methylated between SNORD116 MD neurons and control neurons. Among the differentially methylated genes, we determined a list of genes also differentially expressed. Enrichment analysis of this list encompassed the dopaminergic system with COMT and SLC6A3. COMT displayed hypermethylation and under-expression in SNORD116 MD, and SLC6A3 displayed hypomethylation and over-expression in SNORD116 MD. RT-qPCR confirmed significant over-expression of SLC6A3 in SNORD116 MD neurons. Moreover, the expression of this gene was significantly decreased in the case of OXT adjunction during the differentiation. Conclusion: SNORD116 MD dopaminergic neurons displayed differential methylation and expression in the COMT and SLC6A3 genes, which are related to dopaminergic clearance. |
Portillo, D. Caillet; Puechal, X.; Masson, M.; Kostine, M.; Michaut, A.; Ramon, A.; Wendling, D.; Costedoat-Chalumeau, N.; Richette, P.; Marotte, H.; Vix-Portet, J.; Dubost, J. J.; Ottaviani, S.; Mouterde, G.; Grasland, A.; Frazier, A.; Germain, V.; Coury, F.; Tournadre, A.; Soubrier, M.; Cavalie, L.; Brevet, P.; Zabraniecki, L.; Jamard, B.; Couture, G.; Arnaud, L.; Richez, C.; Degboe, Y.; Ruyssen-Witrand, A.; Constantin, A. Diagnosis and treatment of Tropheryma whipplei infection in patients with inflammatory rheumatic disease: Data from the French Tw-IRD registry Article de journal Dans: J Infect, vol. 88, no. 2, p. 132-138, 2024, ISSN: 1532-2742 (Electronic) 0163-4453 (Linking), (Caillet Portillo, Damien Puechal, Xavier Masson, Maeva Kostine, Marie Michaut, Alexia Ramon, Andre Wendling, Daniel Costedoat-Chalumeau, Nathalie Richette, Pascal Marotte, Hubert Vix-Portet, Justine Dubost, Jean-Jacques Ottaviani, Sebastien Mouterde, Gael Grasland, Anne Frazier, Aline Germain, Vincent Coury, Fabienne Tournadre, Anne Soubrier, Martin Cavalie, Laurent Brevet, Pauline Zabraniecki, Laurent Jamard, Benedicte Couture, Guillaume Arnaud, Laurent Richez, Christophe Degboe, Yannick Ruyssen-Witrand, Adeline Constantin, Arnaud eng England 2023/12/24 J Infect. 2024 Feb;88(2):132-138. doi: 10.1016/j.jinf.2023.12.010. Epub 2023 Dec 22.). @article{RN2123,OBJECTIVES: Tropheryma whipplei infection can manifest as inflammatory joint symptoms, which can lead to misdiagnosis of inflammatory rheumatic disease and the use of disease-modifying antirheumatic drugs. We investigated the impact of diagnosis and treatment of Tropheryma whipplei infection in patients with inflammatory rheumatic disease. METHODS: We initiated a registry including patients with disease-modifying antirheumatic drugs-treated inflammatory rheumatic disease who were subsequently diagnosed with Tropheryma whipplei infection. We collected clinical, biological, treatment data of the inflammatory rheumatic disease, of Tropheryma whipplei infection, and impact of antibiotics on the evolution of inflammatory rheumatic disease. RESULTS: Among 73 inflammatory rheumatic disease patients, disease-modifying antirheumatic drugs initiation triggered extra-articular manifestations in 27% and resulted in stabilisation (51%), worsening (34%), or improvement (15%) of inflammatory rheumatic disease. At the diagnosis of Tropheryma whipplei infection, all patients had rheumatological symptoms (mean age 58 years, median inflammatory rheumatic disease duration 79 months), 84% had extra-rheumatological manifestations, 93% had elevated C-reactive protein, and 86% had hypoalbuminemia. Treatment of Tropheryma whipplei infection consisted mainly of doxycycline plus hydroxychloroquine, leading to remission of Tropheryma whipplei infection in 79% of cases. Antibiotic treatment of Tropheryma whipplei infection was associated with remission of inflammatory rheumatic disease in 93% of cases and enabled disease-modifying antirheumatic drugs and glucocorticoid discontinuation in most cases. CONCLUSIONS: Tropheryma whipplei infection should be considered in inflammatory rheumatic disease patients with extra-articular manifestations, elevated C-reactive protein, and/or hypoalbuminemia before disease-modifying antirheumatic drugs initiation or in inflammatory rheumatic disease patients with an inadequate response to one or more disease-modifying antirheumatic drugs. Positive results of screening and diagnostic tests for Tropheryma whipplei infection involve antibiotic treatment, which is associated with complete recovery of Tropheryma whipplei infection and rapid remission of inflammatory rheumatic disease, allowing disease-modifying antirheumatic drugs and glucocorticoid discontinuation. |
Degboe, Y.; Severino-Freire, M.; Couture, G.; Apoil, P. A.; Gaudenzio, N.; Hermine, O.; Ruyssen-Witrand, A.; Paul, C.; Laroche, M.; Constantin, A.; Livideanu, C. B. The Prevalence Of Osteoporosis Is Low in Adult Cutaneous Mastocytosis Patients Article de journal Dans: J Allergy Clin Immunol Pract, vol. 12, no. 5, p. 1306-1312, 2024, ISSN: 2213-2201 (Electronic), (Degboe, Yannick Severino-Freire, Maella Couture, Guillaume Apoil, Pol-Andre Gaudenzio, Nicolas Hermine, Olivier Ruyssen-Witrand, Adeline Paul, Carle Laroche, Michel Constantin, Arnaud Livideanu, Cristina Bulai eng Research Support, Non-U.S. Gov't 2024/03/01 J Allergy Clin Immunol Pract. 2024 May;12(5):1306-1312. doi: 10.1016/j.jaip.2024.02.021. Epub 2024 Feb 27.). @article{RN2122,BACKGROUND: Systemic mastocytosis (SM) is a clonal disorder of mast cells (MCs) frequently associated with vertebral osteoporosis (OP) and subsequent vertebral fractures (VFs). The natural history of this OP remains unclear. Importantly, we do not know whether OP represents an early event triggered alongside MC abnormalities, and whether MC clonality is sufficient to trigger osteoporosis. OBJECTIVE: To describe OP in patients with medullar clonality in cutaneous mastocytosis (CM) and monoclonal mast cell activation syndrome (MMAS) and to compare their osteoporosis characteristics with those of nonadvanced SM patients (bone marrow mastocytosis and indolent systemic mastocytosis). METHODS: We retrospectively analyzed clinical, biological, and densitometric data of 27 CM, 13 MMAS, and 135 SM patients from the Mastocytosis Expert Center (CEREMAST) in Toulouse, France. RESULTS: The OP (respectively 3.7, 30.8, and 34.1%) and VFs (0.0%, 15.4%, and 20%) were less frequent in CM than in MMAS and SM, despite the presence of clonal MCs in the bone marrow. Most patients with OP and VFs in the non-SM groups had the usual risk factors for OP. Interestingly, the only non-SM patient with a typical SM-like OP had high bone marrow tryptase, developed bone marrow KIT mutation during follow-up, and had a family history of SM. Our data show that OP is not a common clinical finding in CM but is frequent in MMAS. When OP and VFs occur in CM and MMAS patients, they differ from the usual phenotype of SM bone fragility. CONCLUSIONS: Our findings suggest that, in most CM patients, the meaning and management of OP differs from that of OP in MMAS and nonadvanced SM. Prospective longitudinal studies and the validation of predictors are needed to identify CM and MMAS patients developing SM-related OP. |
Briand-Mésange, Fabienne; Gennero, Isabelle; Salles, Juliette; Trudel, Stéphanie; Dahan, Lionel; Ausseil, Jérôme; Payrastre, Bernard; Salles, Jean Pierre; Chap, Hugues From Classical to Alternative Pathways of 2-Arachidonoylglycerol Synthesis: AlterAGs at the Crossroad of Endocannabinoid and Lysophospholipid Signaling Article de journal Dans: Molecules, vol. 29, no. 15, 2024, ISSN: 14203049. @article{Briand-Mesange2024,2-arachidonoylglycerol (2-AG) is the most abundant endocannabinoid (EC), acting as a full agonist at both CB1 and CB2 cannabinoid receptors. It is synthesized on demand in postsynaptic membranes through the sequential action of phosphoinositide-specific phospholipase Cβ1 (PLCβ1) and diacylglycerol lipase α (DAGLα), contributing to retrograde signaling upon interaction with presynaptic CB1. However, 2-AG production might also involve various combinations of PLC and DAGL isoforms, as well as additional intracellular pathways implying other enzymes and substrates. Three other alternative pathways of 2-AG synthesis rest on the extracellular cleavage of 2-arachidonoyl-lysophospholipids by three different hydrolases: glycerophosphodiesterase 3 (GDE3), lipid phosphate phosphatases (LPPs), and two members of ecto-nucleotide pyrophosphatase/phosphodiesterases (ENPP6–7). We propose the names of AlterAG-1, -2, and -3 for three pathways sharing an ectocellular localization, allowing them to convert extracellular lysophospholipid mediators into 2-AG, thus inducing typical signaling switches between various G-protein-coupled receptors (GPCRs). This implies the critical importance of the regioisomerism of both lysophospholipid (LPLs) and 2-AG, which is the object of deep analysis within this review. The precise functional roles of AlterAGs are still poorly understood and will require gene invalidation approaches, knowing that both 2-AG and its related lysophospholipids are involved in numerous aspects of physiology and pathology, including cancer, inflammation, immune defenses, obesity, bone development, neurodegeneration, or psychiatric disorders. |
2023 |
Carle, C.; Degboe, Y.; Ruyssen-Witrand, A.; Arleevskaya, M. I.; Clavel, C.; Renaudineau, Y. Characteristics of the (Auto)Reactive T Cells in Rheumatoid Arthritis According to the Immune Epitope Database Article de journal Dans: Int J Mol Sci, vol. 24, no. 5, 2023, ISSN: 1422-0067 (Electronic) 1422-0067 (Linking), (Carle, Caroline Degboe, Yannick Ruyssen-Witrand, Adeline Arleevskaya, Marina I Clavel, Cyril Renaudineau, Yves eng 2023/Societe Francaise de Rhumatologie/ 17-15-01099/Russian Science Foundation/ Review Switzerland 2023/03/12 Int J Mol Sci. 2023 Feb 21;24(5):4296. doi: 10.3390/ijms24054296.). @article{RN2124,T cells are known to be involved in the pathogenesis of rheumatoid arthritis (RA). Accordingly, and to better understand T cells' contribution to RA, a comprehensive review based on an analysis of the Immune Epitope Database (IEDB) was conducted. An immune CD8+ T cell senescence response is reported in RA and inflammatory diseases, which is driven by active viral antigens from latent viruses and cryptic self-apoptotic peptides. RA-associated pro-inflammatory CD4+ T cells are selected by MHC class II and immunodominant peptides, which are derived from molecular chaperones, host extra-cellular and cellular peptides that could be post-translationally modified (PTM), and bacterial cross-reactive peptides. A large panel of techniques have been used to characterize (auto)reactive T cells and RA-associated peptides with regards to their interaction with the MHC and TCR, capacity to enter the docking site of the shared epitope (DRB1-SE), capacity to induce T cell proliferation, capacity to select T cell subsets (Th1/Th17, Treg), and clinical contribution. Among docking DRB1-SE peptides, those with PTM expand autoreactive and high-affinity CD4+ memory T cells in RA patients with an active disease. Considering original therapeutic options in RA, mutated, or altered peptide ligands (APL) have been developed and are tested in clinical trials. |
2022 |
Rouleau, Coline; Malorie, Margaux; Collet, Corinne; Porquet-Bordes, Valérie; Gennero, Isabelle; Eddiry, Sanaa; Laroche, Michel; Salles, Jean Pierre; Couture, Guillaume; Edouard, Thomas Diagnostic yield of bone fragility gene panel sequencing in children and young adults referred for idiopathic primary osteoporosis at a single regional reference centre Article de journal Dans: Bone Reports, vol. 16, no. February, p. 1–8, 2022, ISSN: 23521872. @article{Rouleau2022,Aim: To describe the presenting features, bone characteristics and molecular genetics in a large monocentric cohort of children and young adults with idiopathic primary osteoporosis. Methods: Sixty-six patients (19 children, 47 adults; 28 males, 38 females; age at referral: 3.8 to 65 years) diagnosed with primary osteoporosis were included in this study; patients with features of osteogenesis imperfecta or other known syndromes associated with osteoporosis were excluded. For each patient, the following data were collected by retrospective chart review: family and personal history of fracture and osteoporosis, mineral homeostasis parameters and markers of bone formation and resorption, bone mineral density (BMD) of the lumbar spine (LS-BMD), the total body less head (TB-BMD), and total hip levels (TH-BMD) measured by DXA. As part of the initial assessment process, a bone fragility gene panel sequencing was performed in all of these patients. Results: There was a higher predominance of males in the children (63%) and of females in the adults (66%) (p = 0.030). Compared to the adults, the children had a significantly lower frequency of vertebral fractures (26 vs 57% |
Bloudeau, Louisa; Linglart, Agnès; Flammier, Sacha; Portefaix, · Aurélie; Bertholet-Thomas, · Aurélia; Eddiry, · Sanaa; Barosi, Anna; Salles, Jean-Pierre; Porquet-Bordes, · Valérie; Rothenbuhler, Anya; Roger, · Christelle; Bacchetta, Justine X-linked hypophosphatemia, obesity and arterial hypertension: data from the XLH21 study FGF21 · FGF23 · Klotho · Obesity · X-linked hypophosphatemia Article de journal Dans: Pediatric Nephrology, vol. 1, p. 3, 2022, ISBN: 0123456789. @article{Bloudeau2022,Background The underlying mechanisms of obesity in X-linked hypophosphatemia (XLH) are not known. We aimed to evaluate whether FGF21, an endocrine FGF involved in the regulation of carbohydrate-lipid metabolism, could be involved. Methods We performed a prospective multicenter cross-sectional study comparing FGF23, Klotho, and FGF21 levels in teenagers with XLH compared to healthy controls (VITADOS cohort) after matching for age, gender, and puberty. Non-parametric tests were performed (results presented as median (min-max)). Results A total of 40 XLH teenagers (n = 20 Standard Of Care, SOC |
Degboe, Y.; Poupot, R.; Poupot, M. Repolarization of Unbalanced Macrophages: Unmet Medical Need in Chronic Inflammation and Cancer Article de journal Dans: Int J Mol Sci, vol. 23, no. 3, 2022, ISSN: 1422-0067 (Electronic) 1422-0067 (Linking), (Degboe, Yannick Poupot, Remy Poupot, Mary eng Review Switzerland 2022/02/16 Int J Mol Sci. 2022 Jan 28;23(3):1496. doi: 10.3390/ijms23031496.). @article{RN2126,Monocytes and their tissue counterpart macrophages (MP) constitute the front line of the immune system. Indeed, they are able to rapidly and efficiently detect both external and internal danger signals, thereby activating the immune system to eradicate the disturbing biological, chemical, or physical agents. They are also in charge of the control of the immune response and account for the repair of the damaged tissues, eventually restoring tissue homeostasis. The balance between these dual activities must be thoroughly controlled in space and time. Any sustained unbalanced response of MP leads to pathological disorders, such as chronic inflammation, or favors cancer development and progression. In this review, we take advantage of our expertise in chronic inflammation, especially in rheumatoid arthritis, and in cancer, to highlight the pivotal role of MP in the physiopathology of these disorders and to emphasize the repolarization of unbalanced MP as a promising therapeutic strategy to control these diseases. |
2021 |
Tauber, Maithé; Hoybye, Charlotte Endocrine disorders in Prader-Willi syndrome: a model to understand and treat hypothalamic dysfunction Article de journal Dans: The Lancet Diabetes and Endocrinology, vol. 9, no. 4, p. 235–246, 2021, ISSN: 22138595. @article{Tauber2021b,Prader-Willi syndrome is a rare genetic neurodevelopmental disorder resulting from the loss of expression of maternally imprinted genes located in the paternal chromosomal region, 15q11–13. Impaired hypothalamic development and function is the cause of most of the phenotypes comprising the developmental trajectory of Prader-Willi syndrome: from anorexia at birth to excessive weight gain preceding hyperphagia, and early severe obesity with hormonal deficiencies, behavioural problems, and dysautonomia. Growth hormone deficiency, hypogonadism, hypothyroidism, premature adrenarche, corticotropin deficiency, precocious puberty, and glucose metabolism disorders are the main endocrine dysfunctions observed. Additionally, as a result of hypothalamic dysfunction, oxytocin and ghrelin systems are impaired in most patients. Standard pituitary and gonadal hormone replacement therapies are required. In this Review, we discuss Prader-Willi syndrome as a model of hypothalamic dysfunction, and provide a comprehensive description of the accumulated knowledge on genetics, pathophysiology, and treatment approaches of this rare disorder. |
Salles, Juliette; Lacassagne, Emmanuelle; Eddiry, Sanaa; Franchitto, Nicolas; Salles, Jean Pierre; Tauber, Maithé What can we learn from PWS and SNORD116 genes about the pathophysiology of addictive disorders? Article de journal Dans: Molecular Psychiatry, vol. 26, no. 1, p. 51–59, 2021, ISSN: 14765578. @article{Salles2021b,Addictive disorders have been much investigated and many studies have underlined the role of environmental factors such as social interaction in the vulnerability to and maintenance of addictive behaviors. Research on addiction pathophysiology now suggests that certain behavioral disorders are addictive, one example being food addiction. Yet, despite the growing body of knowledge on addiction, it is still unknown why only some of the individuals exposed to a drug become addicted to it. This observation has prompted the consideration of genetic heritage, neurodevelopmental trajectories, and gene-environment interactions in addiction vulnerability. Prader–Willi syndrome (PWS) is a rare neurodevelopmental disorder in which children become addicted to food and show early social impairment. PWS is caused by the deficiency of imprinted genes located on the 15q11–q13 chromosome. Among them, the SNORD116 gene was identified as the minimal gene responsible for the PWS phenotype. Several studies have also indicated the role of the Snord116 gene in animal and cellular models to explain PWS pathophysiology and phenotype (including social impairment and food addiction). We thus present here the evidence suggesting the potential involvement of the SNORD116 gene in addictive disorders. |
Eddiry, Sanaa; Diene, Gwenaelle; Molinas, Catherine; Salles, Juliette; Auriol, Françoise Conte; Gennero, Isabelle; Bieth, Eric; Skryabin, Boris V.; Rozhdestvensky, Timofey S.; Burnett, Lisa C.; Leibel, Rudolph L.; Tauber, Maithé; Salles, Jean Pierre SNORD116 and growth hormone therapy impact IGFBP7 in Prader–Willi syndrome Article de journal Dans: Genetics in Medicine, p. 1–9, 2021, ISSN: 1098-3600. @article{Eddiry2021, |
Degboe, Y.; Sunzini, F.; Sood, S.; Bozec, A.; Sokolova, M. V.; Zekovic, A.; McInnes, I. B.; Schett, G.; Goodyear, C. S. Apremilast inhibits inflammatory osteoclastogenesis Article de journal Dans: Rheumatology (Oxford), vol. 61, no. 1, p. 452-461, 2021, ISSN: 1462-0332 (Electronic) 1462-0324 (Linking), (Degboe, Yannick Sunzini, Flavia Sood, Shatakshi Bozec, Aline Sokolova, Maria V Zekovic, Ana McInnes, Iain B Schett, Georg Goodyear, Carl S eng Research Support, Non-U.S. Gov't England 2021/04/01 Rheumatology (Oxford). 2021 Dec 24;61(1):452-461. doi: 10.1093/rheumatology/keab315.). @article{RN2128,OBJECTIVES: Psoriatic arthritis (PsA) is associated with bone erosion and inflammation-induced bone loss, which are mediated by osteoclasts (OC) and modulated by inflammatory cytokines. Apremilast (APR) (a selective phosphodiesterase 4 inhibitor) is efficacious in PsA and acts by inhibiting cytokine production. However, there are no direct data informing whether and how APR affects osteoclast formation in humans. METHODS: Osteoclastogenic cytokine production by activated human peripheral blood mononuclear cells (PBMCs) was measured in the presence and absence of APR. Effects of APR on osteoclast differentiation were tested (i) in co-cultures of activated PBMCs and human CD14+ blood monocytes as well as (ii) in CD14+ blood monocytes stimulated with activated-PBMCs supernatant, TNF or IL-17A. Bone resorption was measured on OsteoAssay plates. Effects of APR on ex vivo osteoclast differentiation were compared in PsA, pre-PsA and psoriasis patients, as well as in healthy controls. RESULTS: APR significantly impaired the expression of key osteoclastogenic cytokines in activated PBMCs. Furthermore, APR dose-dependently and significantly inhibited activated PBMC-driven osteoclast differentiation and ex vivo osteoclast differentiation of PBMCs derived from PsA and pre-PsA patients, but not from psoriasis patients or healthy controls. TNF and IL-17A-enhanced osteoclastogenesis and osteolytic activity of CD14+ blood monocytes from PsA patients was also significantly inhibited by APR. Finally, APR inhibited expression of the key osteoclast fusion protein dendritic cell-specific transmembrane protein. CONCLUSION: Phosphodiesterase 4 targeting by APR not only inhibits osteoclastogenic cytokine production, but also directly suppresses inflammation-driven osteoclastogenesis. These data provide initial evidence that APR has the potential to provide a direct bone protective effect in PsA. |
Diallo, K.; Simons, N.; Sayegh, S.; Baron, M.; Degboe, Y.; Boyer, J. F.; Kruglov, A.; Nedospasov, S.; Novarino, J.; Aloulou, M.; Fazilleau, N.; Constantin, A.; Cantagrel, A.; Davignon, J. L.; Rauwel, B. Evidence for tmTNF reverse signaling in vivo: Implications for an arginase-1-mediated therapeutic effect of TNF inhibitors during inflammation Article de journal Dans: iScience, vol. 24, no. 4, p. 102331, 2021, ISSN: 2589-0042 (Electronic) 2589-0042 (Linking), (Diallo, Katy Simons, Numa Sayegh, Souraya Baron, Michel Degboe, Yannick Boyer, Jean-Frederic Kruglov, Andrey Nedospasov, Sergei Novarino, Julien Aloulou, Meryem Fazilleau, Nicolas Constantin, Arnaud Cantagrel, Alain Davignon, Jean-Luc Rauwel, Benjamin eng 2021/04/24 iScience. 2021 Mar 21;24(4):102331. doi: 10.1016/j.isci.2021.102331. eCollection 2021 Apr 23.). @article{RN2127,In order to ascertain the significance of transmembrane tumor necrosis factor (tmTNF) reverse signaling in vivo, we generated a triple transgenic mouse model (3TG, TNFR1-/-, TNFR2-/-, and tmTNFKI/KI) in which all canonical tumor necrosis factor (TNF) signaling was abolished. In bone-marrow-derived macrophages harvested from these mice, various anti-TNF biologics induced the expression of genes characteristic of alternative macrophages and also inhibited the expression of pro-inflammatory cytokines mainly through the upregulation of arginase-1. Injections of TNF inhibitors during arthritis increased pro-resolutive markers in bone marrow precursors and joint cells leading to a decrease in arthritis score. These results demonstrate that the binding of anti-TNF biologics to tmTNF results in decreased arthritis severity. Collectively, our data provide evidence for the significance of tmTNF reverse signaling in the modulation of arthritis. They suggest a complementary interpretation of anti-TNF biologics effects in the treatment of inflammatory diseases and pave the way to studies focused on new arginase-1-dependent therapeutic targets. |
Degboe, Y. Pre-rheumatoid arthritis and ACPA: Contribution of ACPAs in the pathogeny of pre-disease stage Article de journal Dans: Joint Bone Spine, vol. 88, no. 3, p. 105098, 2021, ISSN: 1778-7254 (Electronic) 1297-319X (Linking), (Degboe, Yannick eng Editorial France 2020/11/07 Joint Bone Spine. 2021 May;88(3):105098. doi: 10.1016/j.jbspin.2020.105098. Epub 2020 Nov 3.). @article{RN2129, |
Salles, Juliette; Briand-Mésange, Fabienne; Trudel, Stephanie; Ausseil, Jérôme; Salles, Jean-Pierre; Chap, Hugues Can antidepressants unlock prescription of rimonabant in the fight against COVID-19? Article de journal Dans: Molecular psychiatry, p. 1–2, 2021. @article{lens.org/045-870-173-132-258, |
2020 |
Briand-Mésange, Fabienne; Pons, Véronique; Allart, Sophie; Masquelier, Julien; Chicanne, Gaëtan; Beton, Nicolas; Payrastre, Bernard; Muccioli, Giulio G.; Ausseil, Jérôme; Davignon, Jean Luc; Salles, Jean Pierre; Chap, Hugues Glycerophosphodiesterase 3 (GDE3) is a lysophosphatidylinositol-specific ectophospholipase C acting as an endocannabinoid signaling switch Article de journal Dans: Journal of Biological Chemistry, vol. 295, no. 46, p. 15767–15781, 2020, ISSN: 1083351X. @article{Briand-Mesange2020,Endocannabinoid signaling plays a regulatory role in various (neuro)biological functions. 2-arachidonoylglycerol (2-AG) is the most abundant endocannabinoid, and although its canonical biosynthetic pathway involving phosphoinositide-specific phospholipase C and diacylglycerol lipase a is known, alternative pathways remain unsettled. Here, we characterize a non-canonical pathway implicating glycerophosphodiesterase 3 (GDE3, from GDPD2 gene). Human GDE3 expressed in HEK293T cell membranes catalyzed the conversion of lysophosphatidylinositol (LPI) into monoacylglycerol and inositol-1-phosphate. The enzyme was equally active against 1-acyl and 2-acyl LPI. When using 2-acyl LPI, where arachidonic acid is the predominant fatty acid, LC-MS analysis identified 2-AG as the main product of LPI hydrolysis by GDE3. Furthermore, inositol-1-phosphate release into the medium occurred upon addition of LPI to intact cells, suggesting that GDE3 is actually an ecto-lysophospholipase C. In cells expressing G-protein–coupled receptor GPR55, GDE3 abolished 1-acyl LPI–induced signaling. In contrast, upon simultaneous expression of GDE3 and cannabinoid receptor CB2, 2-acyl LPI evoked the same signal as that induced by 2-AG. These data strongly suggest that, in addition to degrading the GPR55 LPI ligand, GDE3 can act as a switch between GPR55 and CB2 signaling. Coincident with a major expression of both GDE3 and CB2 in the spleen, spleens from transgenic mice lacking GDE3 displayed doubling of LPI content compared with WT mice. Decreased production of 2-AG in whole spleen was also observed, supporting the in vivo relevance of our findings. These data thus open a new research avenue in the field of endocannabinoid generation and reinforce the view of GPR55 and LPI being genuine actors of the endocannabinoid system. |
Briand-Mésange, Fabienne; Trudel, Stéphanie; Salles, Juliette; Ausseil, Jérôme; Salles, Jean Pierre; Chap, Hugues Possible Role of Adipose Tissue and the Endocannabinoid System in Coronavirus Disease 2019 Pathogenesis: Can Rimonabant Return? Article de journal Dans: Obesity, vol. 28, no. 9, p. 1580–1581, 2020, ISSN: 1930739X. @article{Briand-Mesange2020b, |
Couture, G.; Degboe, Y.; Baujat, G.; Cormier-Daire, V.; Laroche, M. Juvenile osteoporosis with calvarial doughnuts: Progressive high-turnover bone loss responsive to bisphosphonate therapy Article de journal Dans: Joint Bone Spine, vol. 87, no. 3, p. 271-272, 2020, ISSN: 1778-7254 (Electronic) 1297-319X (Linking). @article{RN999, |
Couture, G.; Degboe, Y.; Geniez, C.; Tack, I.; Vallet, M.; Laroche, M. Comment on: Diagnosis of fibromyalgia: comparison of the 2011/2016 ACR and AAPT criteria and validation of the modified Fibromyalgia Assessment Status Article de journal Dans: Rheumatology (Oxford), vol. 59, no. 10, p. e79-e80, 2020, ISSN: 1462-0332 (Electronic) 1462-0324 (Linking). @article{RN998, |
Degboe, Y. Pre-rheumatoid arthritis and ACPA: Contribution of ACPAs in the pathogeny of pre-disease stage Article de journal Dans: Joint Bone Spine, vol. 88, no. 3, p. 105098, 2020, ISSN: 1778-7254 (Electronic) 1297-319X (Linking). @article{RN997, |
Degboe, Y.; Schiff, M.; Weinblatt, M.; Fleischmann, R.; Ahmad, H. A.; Constantin, A. Background Glucocorticoid Therapy Has No Impact on Efficacy and Safety of Abatacept or Adalimumab in Patients with Rheumatoid Arthritis Article de journal Dans: J Clin Med, vol. 9, no. 6, 2020, ISSN: 2077-0383 (Print) 2077-0383 (Linking). @article{RN996, |
Ferrieres, L.; Degboe, Y.; Laroche, M.; Constantin, A.; Ruyssen-Witrand, A. No impact of anti-Rank ligand and PTH analogs on cardiovascular risk in postmenopausal osteoporosis: a systematic literature review and meta-analysis Article de journal Dans: Arch Osteoporos, vol. 15, no. 1, p. 10, 2020, ISSN: 1862-3514 (Electronic). @article{RN995, |
Garcia, P.; Revet, A.; Yrondi, A.; Rousseau, V.; Degboe, Y.; Montastruc, F. Psychiatric Disorders and Hydroxychloroquine for Coronavirus Disease 2019 (COVID-19): A VigiBase Study Article de journal Dans: Drug Saf, vol. 43, no. 12, p. 1315-1322, 2020, ISSN: 1179-1942 (Electronic) 0114-5916 (Linking). @article{RN994, |
Laroche, M.; Champs, B.; Couture, G.; Degboe, Y. Consequence of vertebral fracture cascades: about a cohort of 79 patients Article de journal Dans: Osteoporos Int, vol. 31, no. 12, p. 2497-2498, 2020, ISSN: 1433-2965 (Electronic) 0937-941X (Linking). @article{RN993, |
Laroche, M.; Couture, G.; Ruyssen-Witrand, A.; Constantin, A.; Degboe, Y. Effect of risedronate on bone loss at discontinuation of denosumab Article de journal Dans: Bone Rep, vol. 13, p. 100290, 2020, ISSN: 2352-1872 (Print) 2352-1872 (Linking). @article{RN992, |
Rauwel, B.; Degboe, Y.; Diallo, K.; Sayegh, S.; Baron, M.; Boyer, J. F.; Constantin, A.; Cantagrel, A.; Davignon, J. L. Inhibition of Osteoclastogenesis by the RNA-Binding Protein QKI5: a Novel Approach to Protect from Bone Resorption Article de journal Dans: J Bone Miner Res, vol. 35, no. 4, p. 753-765, 2020, ISSN: 1523-4681 (Electronic) 0884-0431 (Linking). @article{RN991, |
Rauwel, B.; Degboe, Y.; Nigon, D.; Boyer, J. F.; Abravanel, F.; Izopet, J.; Combe, B.; Ruyssen-Witrand, A.; Constantin, A.; Cantagrel, A.; Davignon, J. L. Reduced progression of bone erosion in cytomegalovirus seropositive rheumatoid arthritis patients Article de journal Dans: Arthritis Res Ther, vol. 22, no. 1, p. 13, 2020, ISSN: 1478-6362 (Electronic) 1478-6354 (Linking). @article{RN990, |
Simons, N.; Degboe, Y.; Barnetche, T.; Cantagrel, A.; Ruyssen-Witrand, A.; Constantin, A. Biological DMARD efficacy in psoriatic arthritis: a systematic literature review and meta-analysis on articular, enthesitis, dactylitis, skin and functional outcomes Article de journal Dans: Clin Exp Rheumatol, vol. 38, no. 3, p. 508-515, 2020, ISSN: 0392-856X (Print) 0392-856X (Linking). @article{RN989, |
Vinson, D.; Molet-Benhamou, L.; Degboe, Y.; den Broeder, A.; Ibrahim, F.; Pontes, C.; Westhovens, R.; Zavada, J.; Pham, T.; Barnetche, T.; Constantin, A.; Ruyssen-Witrand, A. Dans: Arthritis Res Ther, vol. 22, no. 1, p. 97, 2020, ISSN: 1478-6362 (Electronic) 1478-6354 (Linking). @article{RN988, |
Kraus, Francois; Dremaux, Julie; Altakfi, Wajd; Goux, Magalie; Pontois, Léa; Sevestre, Henri; Trudel, Stéphanie FOXL2 homozygous genotype and chromosome instability are associated with recurrence in adult granulosa cell tumors of the ovary Article de journal Dans: Oncotarget, vol. 11, no. 4, p. 419–428, 2020, ISSN: 1949-2553. @article{kraus_foxl2_2020,INTRODUCTION: Adult granulosa cell tumors (aGCTs) are extremely rare tumors characterized by the presence of the single missense mutation (c.402 CtextgreaterG, p. C134W) in the FOXL2 gene. These tumors are frequently associated with a slow, indolent disease progression and a high probability of aggressive tumor recurrence. Hence, the identification of molecular markers that are predictive of recurrence and/or aggressive behavior would be a great asset in the management of aGCT. The present study focused on the influence of the FOXL2 genotype (heterozygous or homozygous) and copy number variations (CNVs) in recurrence by comparing the primary tumor with recurrent lesions in the same patient. We performed array comparative genomic hybridization (CGH) experiments and FOXL2 genotyping by allelic discrimination on 40 tumor samples. RESULTS AND DISCUSSION: In array CGH results of recurrent tumors, few samples presented the multiple chromosome losses and gains characteristic of chromosome instability (CIN). We also observed that three recurrent tumors and one primary tumor appeared to be homozygous for the FOXL2 c.402CtextgreaterG mutation. Interestingly, the homozygous FOXL2 genotype was correlated with a shorter time to relapse. A change in the FOXL2 genotype in cases of recurrence was correlated with the appearance of CIN. CONCLUSION: Despite the small number of matching primary and recurrent tumors analyzed here, the present study is the first to have shown that the FOXL2 homozygous genotype and CIN are prevalent in recurrent aGCTs. The two mechanisms are probably linked, and both almost certainly have a role in the molecular transformation of aGCTs. |
Mansour, Ali; Darwiche, Walaa; Yaker, Linda; Da Nascimento, Sophie; Gomila, Cathy; Rossi, Claire; Jung, Vincent; Sonnet, Pascal; Kamel, Saïd; Guerrera, Ida Chiara; Boullier, Agnès; Ausseil, Jérôme GFOGER Peptide Modifies the Protein Content of Extracellular Vesicles and Inhibits Vascular Calcification Article de journal Dans: Frontiers in Cell and Developmental Biology, vol. 8, no. November, p. 1–11, 2020, ISSN: 2296634X. @article{Mansour2020,Objective: Vascular calcification (VC) is an active process during which vascular smooth muscle cells (VSMCs) undergo an osteogenic switch and release extracellular vesicles (EVs). In turn, the EVs serve as calcification foci via interaction with type 1 collagen (COL1). We recently showed that a specific, six-amino-acid repeat (GFOGER) in the sequence of COL1 was involved in the latter's interaction with integrins expressed on EVs. Our main objective was to test the GFOGER ability to inhibit VC. Approach: We synthesized the GFOGER peptide and tested its ability to inhibit the inorganic phosphate (Pi)-induced calcification of VSMCs and aortic rings. Using mass spectrometry, we studied GFOGER's effect on the protein composition of EVs released from Pi-treated VSMCs. Results: Calcification of mouse VSMCs (MOVAS-1 cells), primary human VSMCs, and rat aortic rings was lower in the presence of GFOGER than with Pi alone (with relative decreases of 66, 58, and 91%, respectively; p < 0.001 for all) (no effect was observed with the scramble peptide GOERFG). A comparative proteomic analysis of EVs released from MOVAS-1 cells in the presence or absence of Pi highlighted significant differences in EVs' protein content. Interestingly, the expression of some of the EVs' proteins involved in the calcification process (such as osteogenic markers, TANK-binding kinase 1, and casein kinase II) was diminished in the presence of GFOGER peptide (data are available via ProteomeXchange with identifier PXD018169∗). The decrease of osteogenic marker expression observed in the presence of GFOGER was confirmed by q-RT-PCR analysis. Conclusion: GFOGER peptide reduces vascular calcification by modifying the protein content of the subsequently released EVs, in particular by decreasing osteogenicswitching in VSMCs. |
Beauvieux, Marie Christine; Bérard, Annie M.; Aimone-Gastin, Isabelle; Barbé, Françoise; Barguil, Yann; Collin-Chavagnac, Delphine; Delacour, Hervé; Delevallée, Céline; Nivet-Antoine, Valérie; Peoc'h, Katell; Poupon, Carole; Schmitt, François; Piéroni, Laurence; Sapin, Vincent SFBC working group "Biochemical markers of COVID-19" Article de journal Dans: Annales de Biologie Clinique, vol. 78, no. 3, p. 269–277, 2020, ISSN: 19506112. @article{Beauvieux2020,The SARS-CoV-2 virus is responsible for an epidemic disease called COVID-19, which was initially evidenced in Wuhan, China, and spread very rapidly in China and around the world. In France, the first isolated case seems now to be reported in December 2019, stage 3 of the COVID-19 epidemic was triggered on March 14th, the start of the planned containment exit from May 11th. Healthcare services have faced a large influx of patients who may be beyond their capacity to receive and care, particularly in the Large-East and Ile-de-France regions. Some patients show an evolution of the disease never observed before with other coronaviruses and develop in a few days a very important inflammatory reaction, which can lead to death of patients. A working group of the French Society of Clinical Biology (SFBC) was set up with the objective of providing updated information on the current status of the biological prescriptions (focusing on biochemistry ones) and their evolution during the epidemic, and of analyzing the biological parameters associated with comorbidities and patient evolution in order to link biological results with medical events. The expanded working group covers all sectors of medical biology in France and extends to the French-speaking world: hospital sectors (CHU and CH, Army Training Hospitals) and the private sector opening a field of view on the biological situation in establishments for dependent elderly, social establishments and clinical medical institutions. The purpose of this article is the presentation of this working group and its immediate and future actions. |
Hocquemiller, Michaël; Hemsley, Kim M.; Douglass, Meghan L.; Tamang, Sarah J.; Neumann, Daniel; King, Barbara M.; Beard, Helen; Trim, Paul J.; Winner, Leanne K.; Lau, Adeline A.; Snel, Marten F.; Gomila, Cathy; Ausseil, Jérôme; Mei, Xin; Giersch, Laura; Plavsic, Mark; Laufer, Ralph AAVrh10 Vector Corrects Disease Pathology in MPS IIIA Mice and Achieves Widespread Distribution of SGSH in Large Animal Brains Article de journal Dans: Molecular Therapy - Methods and Clinical Development, vol. 17, no. June, p. 174–187, 2020, ISSN: 23290501. @article{Hocquemiller2020,Patients with mucopolysaccharidosis type IIIA (MPS IIIA) lack the lysosomal enzyme sulfamidase (SGSH), which is responsible for the degradation of heparan sulfate (HS). Build-up of undegraded HS results in severe progressive neurodegeneration for which there is currently no treatment. The ability of the vector adeno-associated virus (AAV)rh.10-CAG-SGSH (LYS-SAF302) to correct disease pathology was evaluated in a mouse model for MPS IIIA. LYS-SAF302 was administered to 5-week-old MPS IIIA mice at three different doses (8.6E+08, 4.1E+10, and 9.0E+10 vector genomes [vg]/animal) injected into the caudate putamen/striatum and thalamus. LYS-SAF302 was able to dose-dependently correct or significantly reduce HS storage, secondary accumulation of GM2 and GM3 gangliosides, ubiquitin-reactive axonal spheroid lesions, lysosomal expansion, and neuroinflammation at 12 weeks and 25 weeks post-dosing. To study SGSH distribution in the brain of large animals, LYS-SAF302 was injected into the subcortical white matter of dogs (1.0E+12 or 2.0E+12 vg/animal) and cynomolgus monkeys (7.2E+11 vg/animal). Increases of SGSH enzyme activity of at least 20% above endogenous levels were detected in 78% (dogs 4 weeks after injection) and 97% (monkeys 6 weeks after injection) of the total brain volume. Taken together, these data validate intraparenchymal AAV administration as a promising method to achieve widespread enzyme distribution and correction of disease pathology in MPS IIIA. |
Yaker, Linda; Kamel, Saïd; Ausseil, Jérôme; Boullier, Agnès Effects of Chronic Kidney Disease and Uremic Toxins on Extracellular Vesicle Biology Article de journal Dans: Toxins, vol. 12, no. 12, p. 1–28, 2020, ISSN: 20726651. @article{Yaker2020,Vascular calcification (VC) is a cardiovascular complication associated with a high mortality rate, especially in patients with diabetes, atherosclerosis or chronic kidney disease (CKD). In CKD patients, VC is associated with the accumulation of uremic toxins, such as indoxyl sulphate or inorganic phosphate, which can have a major impact in vascular remodeling. During VC, vascular smooth muscle cells (VSMCs) undergo an osteogenic switch and secrete extracellular vesicles (EVs) that are heterogeneous in terms of their origin and composition. Under physiological conditions, EVs are involved in cell-cell communication and the maintenance of cellular homeostasis. They contain high levels of calcification inhibitors, such as fetuin-A and matrix Gla protein. Under pathological conditions (and particularly in the presence of uremic toxins), the secreted EVs acquire a pro-calcifying profile and thereby act as nucleating foci for the crystallization of hydroxyapatite and the propagation of calcification. Here, we review the most recent findings on the EVs' pathophysiological role in VC, the impact of uremic toxins on EV biogenesis and functions, the use of EVs as diagnostic biomarkers and the EVs' therapeutic potential in CKD. |
Blouin, Jean-Marc; Ged, Cécile; Lalanne, Magalie; Lamrissi-Garcia, Isabelle; Morice-Picard, Fanny; Costet, Pierre; Daher, Raêd; Moreau-Gaudry, François; Bedel, Aurélie; Puy, Hervé; Gouya, Laurent; Karim, Zoubida; Richard, Emmanuel Iron chelation rescues hemolytic anemia and skin photosensitivity in congenital erythropoietic porphyria Article de journal Dans: Blood, vol. 136, no. 21, p. 2457–2468, 2020, ISSN: 1528-0020. @article{blouin_iron_2020,Congenital erythropoietic porphyria (CEP) is an inborn error of heme synthesis resulting from uroporphyrinogen III synthase (UROS) deficiency and the accumulation of nonphysiological porphyrin isomer I metabolites. Clinical features are heterogeneous among patients with CEP but usually combine skin photosensitivity and chronic hemolytic anemia, the severity of which is related to porphyrin overload. Therapeutic options include symptomatic strategies only and are unsatisfactory. One promising approach to treating CEP is to reduce the erythroid production of porphyrins through substrate reduction therapy by inhibiting 5-aminolevulinate synthase 2 (ALAS2), the first and rate-limiting enzyme in the heme biosynthetic pathway. We efficiently reduced porphyrin accumulation after RNA interference-mediated downregulation of ALAS2 in human erythroid cellular models of CEP disease. Taking advantage of the physiological iron-dependent posttranscriptional regulation of ALAS2, we evaluated whether iron chelation with deferiprone could decrease ALAS2 expression and subsequent porphyrin production in vitro and in vivo in a CEP murine model. Treatment with deferiprone of UROS-deficient erythroid cell lines and peripheral blood CD34+-derived erythroid cultures from a patient with CEP inhibited iron-dependent protein ALAS2 and iron-responsive element-binding protein 2 expression and reduced porphyrin production. Furthermore, porphyrin accumulation progressively decreased in red blood cells and urine, and skin photosensitivity in CEP mice treated with deferiprone (1 or 3 mg/mL in drinking water) for 26 weeks was reversed. Hemolysis and iron overload improved upon iron chelation with full correction of anemia in CEP mice treated at the highest dose of deferiprone. Our findings highlight, in both mouse and human models, the therapeutic potential of iron restriction to modulate the phenotype in CEP. |
Tybl, Elisabeth; Gunshin, Hiromi; Gupta, Sanjay; Barrientos, Tomasa; Bonadonna, Michael; Celma Nos, Ferran; Palais, Gael; Karim, Zoubida; Sanchez, Mayka; Andrews, Nancy C.; Galy, Bruno Control of Systemic Iron Homeostasis by the 3' Iron-Responsive Element of Divalent Metal Transporter 1 in Mice Article de journal Dans: HemaSphere, vol. 4, no. 5, p. e459, 2020, ISSN: 2572-9241. @article{tybl_control_2020,Supplemental Digital Content is available in the text. |
Touzot, Maxime; Lefebvre, Thibaud; Maheas, Catherine; Ridel, Christophe; Puy, Hervé; Karim, Zoubida A hepcidin-based approach for iron therapy in hemodialysis patients: A pilot study Article de journal Dans: Hemodialysis International. International Symposium on Home Hemodialysis, vol. 24, no. 2, p. 188–194, 2020, ISSN: 1542-4758. @article{touzot_hepcidin-based_2020,INTRODUCTION: Hepcidin is a key factor that regulates iron homeostasis. In hemodialysis patients (HD), a high hepcidin level may decrease intestinal iron absorption and reduce the efficacy of Oral iron vs Intravenous iron therapy. Whether the hepcidin level in HD could guide oral iron therapy is unclear. METHODS: We report a monocentric study on nine "erythropoietin (EPO)-free" patients (without recombinant human EPO [rHU-EPO] for at least 6 months) and normal hepcidin level (textless20 ng mL) during the study. After 15 days of washout, oral iron (ferrous sulfate 80 mg/day) was introduced. The primary end point was the hemoglobin response and iron store at 3 months. FINDINGS: Nine patients (8 men, 1 woman) with a median age of 62 years (range 42-79) were included. After 1 week of treatment, the median transferrin saturation index increased from 15% (range 6-61) to 34% (range 13-42) |
Blouin, Jean-Marc; Ged, Cécile; Lalanne, Magalie; Lamrissi-Garcia, Isabelle; Morice-Picard, Fanny; Costet, Pierre; Daher, Raêd; Moreau-Gaudry, François; Bedel, Aurélie; Puy, Hervé; Gouya, Laurent; Karim, Zoubida; Richard, Emmanuel Iron chelation rescues hemolytic anemia and skin photosensitivity in congenital erythropoietic porphyria Article de journal Dans: Blood, vol. 136, no. 21, p. 2457–2468, 2020, ISSN: 1528-0020. @article{blouin_iron_2020b,Congenital erythropoietic porphyria (CEP) is an inborn error of heme synthesis resulting from uroporphyrinogen III synthase (UROS) deficiency and the accumulation of nonphysiological porphyrin isomer I metabolites. Clinical features are heterogeneous among patients with CEP but usually combine skin photosensitivity and chronic hemolytic anemia, the severity of which is related to porphyrin overload. Therapeutic options include symptomatic strategies only and are unsatisfactory. One promising approach to treating CEP is to reduce the erythroid production of porphyrins through substrate reduction therapy by inhibiting 5-aminolevulinate synthase 2 (ALAS2), the first and rate-limiting enzyme in the heme biosynthetic pathway. We efficiently reduced porphyrin accumulation after RNA interference-mediated downregulation of ALAS2 in human erythroid cellular models of CEP disease. Taking advantage of the physiological iron-dependent posttranscriptional regulation of ALAS2, we evaluated whether iron chelation with deferiprone could decrease ALAS2 expression and subsequent porphyrin production in vitro and in vivo in a CEP murine model. Treatment with deferiprone of UROS-deficient erythroid cell lines and peripheral blood CD34+-derived erythroid cultures from a patient with CEP inhibited iron-dependent protein ALAS2 and iron-responsive element-binding protein 2 expression and reduced porphyrin production. Furthermore, porphyrin accumulation progressively decreased in red blood cells and urine, and skin photosensitivity in CEP mice treated with deferiprone (1 or 3 mg/mL in drinking water) for 26 weeks was reversed. Hemolysis and iron overload improved upon iron chelation with full correction of anemia in CEP mice treated at the highest dose of deferiprone. Our findings highlight, in both mouse and human models, the therapeutic potential of iron restriction to modulate the phenotype in CEP. |
Tybl, Elisabeth; Gunshin, Hiromi; Gupta, Sanjay; Barrientos, Tomasa; Bonadonna, Michael; Celma Nos, Ferran; Palais, Gael; Karim, Zoubida; Sanchez, Mayka; Andrews, Nancy C.; Galy, Bruno Control of Systemic Iron Homeostasis by the 3' Iron-Responsive Element of Divalent Metal Transporter 1 in Mice Article de journal Dans: HemaSphere, vol. 4, no. 5, p. e459, 2020, ISSN: 2572-9241. @article{tybl_control_2020b,Supplemental Digital Content is available in the text. |
Touzot, Maxime; Lefebvre, Thibaud; Maheas, Catherine; Ridel, Christophe; Puy, Hervé; Karim, Zoubida A hepcidin-based approach for iron therapy in hemodialysis patients: A pilot study Article de journal Dans: Hemodialysis International. International Symposium on Home Hemodialysis, vol. 24, no. 2, p. 188–194, 2020, ISSN: 1542-4758. @article{touzot_hepcidin-based_2020b,INTRODUCTION: Hepcidin is a key factor that regulates iron homeostasis. In hemodialysis patients (HD), a high hepcidin level may decrease intestinal iron absorption and reduce the efficacy of Oral iron vs Intravenous iron therapy. Whether the hepcidin level in HD could guide oral iron therapy is unclear. METHODS: We report a monocentric study on nine "erythropoietin (EPO)-free" patients (without recombinant human EPO [rHU-EPO] for at least 6 months) and normal hepcidin level (textless20 ng mL) during the study. After 15 days of washout, oral iron (ferrous sulfate 80 mg/day) was introduced. The primary end point was the hemoglobin response and iron store at 3 months. FINDINGS: Nine patients (8 men, 1 woman) with a median age of 62 years (range 42-79) were included. After 1 week of treatment, the median transferrin saturation index increased from 15% (range 6-61) to 34% (range 13-42) |
Barre, Ronan; Beton, Nicolas; Batut, Aurélie; Accabled, Frank; Sales de Gauzy, Jerome; Auriol, Françoise; Eddiry, Sanaa; Tauber, Maithe; Laurencin, Sara; Salles, Jean Pierre; Gennero, Isabelle Ghrelin uses the GHS-R1a/Gi/cAMP pathway and induces differentiation only in mature osteoblasts. This ghrelin pathway is impaired in AIS patients Article de journal Dans: Biochemistry and Biophysics Reports, vol. 24, 2020, ISSN: 24055808. @article{Barre2020b,We have examined the Acylated Ghrelin (AG)/Gi pathway in different human osteoblastic cell lines. We have found that: 1) AG induces differentiation/mineralization only in mature osteoblasts; 2) the expression of GHS-R1a increases up to the mature cell stage, 3) the action is mediated via the GHS-R/Gi/cAMP pathway only in mature osteoblasts, and 4) osteoblastic cells from adolescent idiopathic scoliosis (AIS) are resistant to the AG/Gi/cAMP pathway. Altogether, these results suggested that AG uses the GHS-R1a/Gi/cAMP pathway to induce differentiation in mature osteoblasts only. This pathway is impaired in AIS osteoblasts. Understanding AG-specific pathways involved in normal and pathological osteoblasts may be useful for developing new treatments for pathologies such as AIS or osteoporosis. |
Salles, Juliette; Lacassagne, Emmanuelle; Benvegnu, Grégoire; Berthoumieu, Sophie Çabal; Franchitto, Nicolas; Tauber, Maithé The RDoC approach for translational psychiatry: Could a genetic disorder with psychiatric symptoms help fill the matrix? the example of Prader–Willi syndrome Article de journal Dans: Translational Psychiatry, vol. 10, no. 1, 2020, ISSN: 21583188. @article{Salles2020b,The Research Domain Criteria project (RDoc) proposes a new classification system based on information from several fields in order to encourage translational perspectives. Nevertheless, integrating genetic markers into this classification has remained difficult because of the lack of powerful associations between targeted genes and RDoC domains. We hypothesized that genetic diseases with psychiatric manifestations would be good models for RDoC gene investigations and would thereby extend the translational approach to involve targeted gene pathways. To explore this possibility, we reviewed the current knowledge on Prader–Willi syndrome, a genetic disorder caused by the absence of expression of some of the genes of the chromosome 15q11–13 region inherited from the father. Indeed, we found that the associations between genes of the PW locus and the modification identified in the relevant behavioral, physiological, and brain imaging studies followed the structure of the RDoC matrix and its six domains (positive valence, negative valence, social processing, cognitive systems, arousal/regulatory systems, and sensorimotor systems). |
Rauwel, B.; Degboe, Y.; Diallo, K.; Sayegh, S.; Baron, M.; Boyer, J. F.; Constantin, A.; Cantagrel, A.; Davignon, J. L. Inhibition of Osteoclastogenesis by the RNA-Binding Protein QKI5: a Novel Approach to Protect from Bone Resorption Article de journal Dans: J Bone Miner Res, vol. 35, no. 4, p. 753-765, 2020, ISSN: 1523-4681 (Electronic) 0884-0431 (Linking), (Rauwel, Benjamin Degboe, Yannick Diallo, Katy Sayegh, Souraya Baron, Michel Boyer, Jean-Frederic Constantin, Arnaud Cantagrel, Alain Davignon, Jean-Luc eng Research Support, Non-U.S. Gov't England 2019/12/14 J Bone Miner Res. 2020 Apr;35(4):753-765. doi: 10.1002/jbmr.3943. Epub 2019 Dec 31.). @article{RN2131,Increased osteoclastogenesis is a common feature of bone erosion, notably in osteoporosis but also in inflammatory diseases such as rheumatoid arthritis (RA) and osteoarticular infections. Human cytomegalovirus (HCMV) infection has been described to impair monocyte differentiation into macrophages and dendritic cells. However, its effect on monocyte-derived osteoclasts is yet to be determined. We showed here that in vitro HCMV infection is associated with an inhibition of osteoclastogenesis through decreased expression of colony stimulating factor 1 receptor (CSF-1R) and RANK in monocytes, which was mediated by an upregulation of quaking I-5 protein (QKI-5), a cellular RNA-interacting protein. We found that deliberate QKI5 overexpression in the absence of HCMV infection is able to decrease CSF-1R and RANK expression, leading to osteoclastogenesis inhibition. Finally, by using lentiviral vectors in a calvarial bone erosion mouse model, we showed that QKI5 inhibits bone degradation. This work identifies QKI5 as a strong inhibitor of bone resorption. Future research will point out whether QKI5 could be a target for bone pathologies. (c) 2019 American Society for Bone and Mineral Research. |
2019 |
Peoc'h, Katell; Nicolas, Gaël; Schmitt, Caroline; Mirmiran, Arienne; Daher, Raed; Lefebvre, Thibaud; Gouya, Laurent; Karim, Zoubida; Puy, Hervé Regulation and tissue-specific expression of δ-aminolevulinic acid synthases in non-syndromic sideroblastic anemias and porphyrias Article de journal Dans: Molecular Genetics and Metabolism, vol. 128, no. 3, p. 190–197, 2019, ISSN: 1096-7206. @article{peoch_regulation_2019,Recently, new genes and molecular mechanisms have been identified in patients with porphyrias and sideroblastic anemias (SA). They all modulate either directly or indirectly the δ-aminolevulinic acid synthase (ALAS) activity. ALAS, is encoded by two genes: the erythroid-specific (ALAS2), and the ubiquitously expressed (ALAS1). In the liver, ALAS1 controls the rate-limiting step in the production of heme and hemoproteins that are rapidly turned over in response to metabolic needs. Several heme regulatory targets have been identified as regulators of ALAS1 activity: 1) transcriptional repression via a heme-responsive element, 2) post-transcriptional destabilization of ALAS1 mRNA, 3) post-translational inhibition via a heme regulatory motif, 4) direct inhibition of the activity of the enzyme and 5) breakdown of ALAS1 protein via heme-mediated induction of the protease Lon peptidase 1. In erythroid cells, ALAS2 is a gatekeeper of production of very large amounts of heme necessary for hemoglobin synthesis. The rate of ALAS2 synthesis is transiently increased during the period of active heme synthesis. Its gene expression is determined by trans-activation of nuclear factor GATA1, CACC box and NF-E2-binding sites in the promoter areas. ALAS2 mRNA translation is also regulated by the iron-responsive element (IRE)/iron regulatory proteins (IRP) binding system. In patients, ALAS enzyme activity is affected in most of the mutations causing non-syndromic SA and in several porphyrias. Decreased ALAS2 activity results either directly from loss-of-function ALAS2 mutations as seen in X-linked sideroblastic anemia (XLSA) or from defect in the availability of one of its two mitochondrial substrates: glycine in SLC25A38 mutations and succinyl CoA in GLRX5 mutations. Moreover, ALAS2 gain of function mutations is responsible for X-linked protoporphyria and increased ALAS1 activity lead to acute attacks of hepatic porphyrias. A missense dominant mutation in the Walker A motif of the ATPase binding site in the gene coding for the mitochondrial protein unfoldase CLPX also contributes to increasing ALAS and subsequently protoporphyrinemia. Altogether, these recent data on human ALAS have informed our understanding of porphyrias and sideroblastic anemias pathogeneses and may contribute to new therapeutic strategies. |
Daher, Raêd; Mansouri, Abdellah; Martelli, Alain; Bayart, Sophie; Manceau, Hana; Callebaut, Isabelle; Moulouel, Boualem; Gouya, Laurent; Puy, Hervé; Kannengiesser, Caroline; Karim, Zoubida GLRX5 mutations impair heme biosynthetic enzymes ALA synthase 2 and ferrochelatase in Human congenital sideroblastic anemia Article de journal Dans: Molecular Genetics and Metabolism, vol. 128, no. 3, p. 342–351, 2019, ISSN: 1096-7206. @article{daher_glrx5_2019,Non-syndromic microcytic congenital sideroblastic anemia (cSA) is predominantly caused by defective genes encoding for either ALAS2, the first enzyme of heme biosynthesis pathway or SLC25A38, the mitochondrial importer of glycine, an ALAS2 substrate. Herein we explored a new case of cSA with two mutations in GLRX5, a gene for which only two patients have been reported so far. The patient was a young female with biallelic compound heterozygous mutations in GLRX5 (p.Cys67Tyr and p.Met128Lys). Three-D structure analysis confirmed the involvement of Cys67 in the coordination of the [2Fe2S] cluster and suggested a potential role of Met128 in partner interactions. The protein-level of ferrochelatase, the terminal-enzyme of heme process, was increased both in patient-derived lymphoblastoid and CD34+ cells, however, its activity was drastically decreased. The activity of ALAS2 was found altered and possibly related to a defect in the biogenesis of its co-substrate, the succinyl-CoA. Thus, the patient exhibits both a very low ferrochelatase activity without any accumulation of porphyrins precursors in contrast to what is reported in erythropoietic protoporphyria with solely impaired ferrochelatase activity. A significant oxidative stress was evidenced by decreased reduced glutathione and aconitase activity, and increased MnSOD protein expression. This oxidative stress depleted and damaged mtDNA, decreased complex I and IV activities and depleted ATP content. Collectively, our study demonstrates the key role of GLRX5 in modulating ALAS2 and ferrochelatase activities and in maintaining mitochondrial function. |
Peoc'h, Katell; Manceau, Hana; Karim, Zoubida; Wahlin, Staffan; Gouya, Laurent; Puy, Hervé; Deybach, Jean-Charles Hepatocellular carcinoma in acute hepatic porphyrias: A Damocles Sword Article de journal Dans: Molecular Genetics and Metabolism, vol. 128, no. 3, p. 236–241, 2019, ISSN: 1096-7206. @article{peoch_hepatocellular_2019,Porphyrias are inherited diseases with low penetrance affecting the heme biosynthesis pathway. Acute intermittent porphyria (AIP), variegate porphyria (VP) and hereditary coproporphyria (HCP) together constitute the acute hepatic porphyrias (AHP). These diseases have been identified as risk factors for primary liver cancers (PLC), mainly hepatocellular carcinoma (HCC: range 87-100%) but also cholangiocarcinoma, alone or combination with HCC. In AHP, HCC annual incidence rates range from 0.16 to 0.35% according to the populations studied. Annual incidence rates are higher in Swedish and Norwegian patients, due to a founder effect. It increases above age 50. The pathophysiology could include both direct toxic effects of heme precursors, particularly δ-aminolevulinic acid (ALA), compound heterozygosity for genes implied in heme biosynthesis pathway or the loss of oxidative stress homeostasis due to a relative lack of heme. The high HCC incidence justifies radiological surveillance in AHP patients above age 50. Efforts are made to find new biological non-invasive markers. In this respect, we describe here the first report of PIVKA-II clinical utility in the follow-up of an AIP patient that develop an HCC. In this manuscript we reviewed the epidemiology, the physiopathology, and the screening strategy of HCC in AHP. |
Peoc'h, Katell; Nicolas, Gaël; Schmitt, Caroline; Mirmiran, Arienne; Daher, Raed; Lefebvre, Thibaud; Gouya, Laurent; Karim, Zoubida; Puy, Hervé Regulation and tissue-specific expression of δ-aminolevulinic acid synthases in non-syndromic sideroblastic anemias and porphyrias Article de journal Dans: Molecular Genetics and Metabolism, vol. 128, no. 3, p. 190–197, 2019, ISSN: 1096-7206. @article{peoch_regulation_2019b,Recently, new genes and molecular mechanisms have been identified in patients with porphyrias and sideroblastic anemias (SA). They all modulate either directly or indirectly the δ-aminolevulinic acid synthase (ALAS) activity. ALAS, is encoded by two genes: the erythroid-specific (ALAS2), and the ubiquitously expressed (ALAS1). In the liver, ALAS1 controls the rate-limiting step in the production of heme and hemoproteins that are rapidly turned over in response to metabolic needs. Several heme regulatory targets have been identified as regulators of ALAS1 activity: 1) transcriptional repression via a heme-responsive element, 2) post-transcriptional destabilization of ALAS1 mRNA, 3) post-translational inhibition via a heme regulatory motif, 4) direct inhibition of the activity of the enzyme and 5) breakdown of ALAS1 protein via heme-mediated induction of the protease Lon peptidase 1. In erythroid cells, ALAS2 is a gatekeeper of production of very large amounts of heme necessary for hemoglobin synthesis. The rate of ALAS2 synthesis is transiently increased during the period of active heme synthesis. Its gene expression is determined by trans-activation of nuclear factor GATA1, CACC box and NF-E2-binding sites in the promoter areas. ALAS2 mRNA translation is also regulated by the iron-responsive element (IRE)/iron regulatory proteins (IRP) binding system. In patients, ALAS enzyme activity is affected in most of the mutations causing non-syndromic SA and in several porphyrias. Decreased ALAS2 activity results either directly from loss-of-function ALAS2 mutations as seen in X-linked sideroblastic anemia (XLSA) or from defect in the availability of one of its two mitochondrial substrates: glycine in SLC25A38 mutations and succinyl CoA in GLRX5 mutations. Moreover, ALAS2 gain of function mutations is responsible for X-linked protoporphyria and increased ALAS1 activity lead to acute attacks of hepatic porphyrias. A missense dominant mutation in the Walker A motif of the ATPase binding site in the gene coding for the mitochondrial protein unfoldase CLPX also contributes to increasing ALAS and subsequently protoporphyrinemia. Altogether, these recent data on human ALAS have informed our understanding of porphyrias and sideroblastic anemias pathogeneses and may contribute to new therapeutic strategies. |
Daher, Raêd; Mansouri, Abdellah; Martelli, Alain; Bayart, Sophie; Manceau, Hana; Callebaut, Isabelle; Moulouel, Boualem; Gouya, Laurent; Puy, Hervé; Kannengiesser, Caroline; Karim, Zoubida GLRX5 mutations impair heme biosynthetic enzymes ALA synthase 2 and ferrochelatase in Human congenital sideroblastic anemia Article de journal Dans: Molecular Genetics and Metabolism, vol. 128, no. 3, p. 342–351, 2019, ISSN: 1096-7206. @article{daher_glrx5_2019b,Non-syndromic microcytic congenital sideroblastic anemia (cSA) is predominantly caused by defective genes encoding for either ALAS2, the first enzyme of heme biosynthesis pathway or SLC25A38, the mitochondrial importer of glycine, an ALAS2 substrate. Herein we explored a new case of cSA with two mutations in GLRX5, a gene for which only two patients have been reported so far. The patient was a young female with biallelic compound heterozygous mutations in GLRX5 (p.Cys67Tyr and p.Met128Lys). Three-D structure analysis confirmed the involvement of Cys67 in the coordination of the [2Fe2S] cluster and suggested a potential role of Met128 in partner interactions. The protein-level of ferrochelatase, the terminal-enzyme of heme process, was increased both in patient-derived lymphoblastoid and CD34+ cells, however, its activity was drastically decreased. The activity of ALAS2 was found altered and possibly related to a defect in the biogenesis of its co-substrate, the succinyl-CoA. Thus, the patient exhibits both a very low ferrochelatase activity without any accumulation of porphyrins precursors in contrast to what is reported in erythropoietic protoporphyria with solely impaired ferrochelatase activity. A significant oxidative stress was evidenced by decreased reduced glutathione and aconitase activity, and increased MnSOD protein expression. This oxidative stress depleted and damaged mtDNA, decreased complex I and IV activities and depleted ATP content. Collectively, our study demonstrates the key role of GLRX5 in modulating ALAS2 and ferrochelatase activities and in maintaining mitochondrial function. |
Peoc'h, Katell; Manceau, Hana; Karim, Zoubida; Wahlin, Staffan; Gouya, Laurent; Puy, Hervé; Deybach, Jean-Charles Hepatocellular carcinoma in acute hepatic porphyrias: A Damocles Sword Article de journal Dans: Molecular Genetics and Metabolism, vol. 128, no. 3, p. 236–241, 2019, ISSN: 1096-7206. @article{peoch_hepatocellular_2019b,Porphyrias are inherited diseases with low penetrance affecting the heme biosynthesis pathway. Acute intermittent porphyria (AIP), variegate porphyria (VP) and hereditary coproporphyria (HCP) together constitute the acute hepatic porphyrias (AHP). These diseases have been identified as risk factors for primary liver cancers (PLC), mainly hepatocellular carcinoma (HCC: range 87-100%) but also cholangiocarcinoma, alone or combination with HCC. In AHP, HCC annual incidence rates range from 0.16 to 0.35% according to the populations studied. Annual incidence rates are higher in Swedish and Norwegian patients, due to a founder effect. It increases above age 50. The pathophysiology could include both direct toxic effects of heme precursors, particularly δ-aminolevulinic acid (ALA), compound heterozygosity for genes implied in heme biosynthesis pathway or the loss of oxidative stress homeostasis due to a relative lack of heme. The high HCC incidence justifies radiological surveillance in AHP patients above age 50. Efforts are made to find new biological non-invasive markers. In this respect, we describe here the first report of PIVKA-II clinical utility in the follow-up of an AIP patient that develop an HCC. In this manuscript we reviewed the epidemiology, the physiopathology, and the screening strategy of HCC in AHP. |
Touzot, Maxime; Lefebvre, Thibaud; Roux, Arthur; Maheas, Catherine; Ridel, Christophe; Puy, Hervé; Karim, Zoubida Functional erythropoietin-hepcidin axis in recombinant human erythropoietin independent haemodialysis patients Article de journal Dans: Nephrology (Carlton, Vic.), vol. 24, no. 7, p. 751–757, 2019, ISSN: 1440-1797. @article{touzot_functional_2019,AIM: Relatively few haemodialysis (HD) patients remain independent of recombinant human erythropoietin ('rHU-EPO free patients'). We investigated the role of EPO and hepcidin, two key hormones involved in anaemia. METHODS: We report a monocentric case-control series. Iron status, EPO and hepcidin levels were analysed in 15 Adult HD (Age textgreater 18 years) with a stable haemoglobin (Hb) level that have not received rHU-EPO for at least 6 months (=rHU-EPO free patients); and in 60 controls with a stable rHU-EPO dose and Hb level. RESULTS: The rHU-EPO free patients had a higher Hb level compared to controls (12.1 ± 0.99 g/dL vs 11.1 ± 0.73, P = 0.0014), and a lower ferritin level (183 ± 102 vs 312 ± 166 ng/mL, P = 0.001). Hepcidin levels were lower in the rHU-EPO free patients (12.53 ± 10.46 ng/mL) compared to the controls (37.95 ± 34.33 ng/mL), P = 0.0033. Hepcidin levels correlated significantly with ferritin levels; but neither with transferrin saturation, C-reactive protein nor EPO levels. Unsupervised analysis revealed that rHU-EPO free patients had a specific clinical/biological profile (presence of renal cyst, longer dialysis vintage, lower ferritin, and EPO and hepcidin levels compared to the control group). Finally, we showed that a lower ferritin level might be a surrogate marker of a lower hepcidin status in this population. CONCLUSION: Recombinant human erythropoietin free patients seem to restore the EPO-hepcidin axis that is critical for erythropoiesis. A specific combination of clinical and biological parameters may help to detect future rHU-EPO free patients. |
Park, Sophie; Kosmider, Olivier; Maloisel, Frédéric; Drenou, Bernard; Chapuis, Nicolas; Lefebvre, Thibaud; Karim, Zoubida; Puy, Hervé; Alary, Anne Sophie; Ducamp, Sarah; Verdier, Frédérique; Bouilloux, Cécile; Rousseau, Alice; Jacob, Marie-Christine; Debliquis, Agathe; Charpentier, Agnes; Gyan, Emmanuel; Anglaret, Bruno; Leyronnas, Cecile; Corm, Selim; Slama, Borhane; Cheze, Stephane; Laribi, Kamel; Amé, Shanti; Rose, Christian; Lachenal, Florence; Toma, Andrea; Pica, Gian Matteo; Carre, Martin; Garban, Frédéric; Mariette, Clara; Cahn, Jean-Yves; Meunier, Mathieu; Herault, Olivier; Fenaux, Pierre; Wagner-Ballon, Orianne; Bardet, Valerie; Dreyfus, Francois; Fontenay, Michaela Dyserythropoiesis evaluated by the RED score and hepcidin:ferritin ratio predicts response to erythropoietin in lower-risk myelodysplastic syndromes Article de journal Dans: Haematologica, vol. 104, no. 3, p. 497–504, 2019, ISSN: 1592-8721. @article{park_dyserythropoiesis_2019,Erythropoiesis-stimulating agents are generally the first line of treatment of anemia in patients with lower-risk myelodysplastic syndrome. We prospectively investigated the predictive value of somatic mutations, and biomarkers of ineffective erythropoiesis including the flow cytometry RED score, serum growth-differentiation factor-15, and hepcidin levels. Inclusion criteria were no prior treatment with erythropoiesis-stimulating agents, low- or intermediate-1-risk myelodysplastic syndrome according to the International Prognostic Scoring System, and a hemoglobin level textless10 g/dL. Patients could be red blood cell transfusion-dependent or not and were given epoetin zeta 40 000 IU/week. Serum erythropoietin level, iron parameters, hepcidin, flow cytometry Ogata and RED scores, and growth-differentiation factor-15 levels were determined at baseline, and molecular analysis by next-generation sequencing was also conducted. Erythroid response (defined according to the International Working Group 2006 criteria) was assessed at week 12. Seventy patients, with a median age of 78 years, were included in the study. There were 22 patients with refractory cytopenia with multilineage dysplasia, 19 with refractory cytopenia with unilineage dysplasia, 14 with refractory anemia with ring sideroblasts, four with refractory anemia with excess blasts-1, six with chronic myelomonocytic leukemia, two with del5q-and three with unclassifiable myelodysplastic syndrome. According to the revised International Prognostic Scoring System, 13 had very low risk, 47 had low risk, nine intermediate risk and one had high-risk disease. Twenty patients were transfusion dependent. Forty-eight percent had an erythroid response and the median duration of the response was 26 months. At baseline, non-responders had significantly higher RED scores and lower hepcidin:ferritin ratios. In multivariate analysis, only a RED score textgreater4 (P=0.05) and a hepcidin:ferritin ratio textless9 (P=0.02) were statistically significantly associated with worse erythroid response. The median response duration was shorter in patients with growth-differentiation factor-15 textgreater2000 pg/mL and a hepcidin:ferritin ratio textless9 (P=0.0008 and P=0.01, respectively). In multivariate analysis, both variables were associated with shorter response duration. Erythroid response to epoetin zeta was similar to that obtained with other erythropoiesis-stimulating agents and was correlated with higher baseline hepcidin:ferritin ratio and lower RED score. ClinicalTrials.gov registration: NCT 03598582. |
Park, Sophie; Kosmider, Olivier; Maloisel, Frédéric; Drenou, Bernard; Chapuis, Nicolas; Lefebvre, Thibaud; Karim, Zoubida; Puy, Hervé; Alary, Anne Sophie; Ducamp, Sarah; Verdier, Frédérique; Bouilloux, Cécile; Rousseau, Alice; Jacob, Marie-Christine; Debliquis, Agathe; Charpentier, Agnes; Gyan, Emmanuel; Anglaret, Bruno; Leyronnas, Cecile; Corm, Selim; Slama, Borhane; Cheze, Stephane; Laribi, Kamel; Amé, Shanti; Rose, Christian; Lachenal, Florence; Toma, Andrea; Pica, Gian Matteo; Carre, Martin; Garban, Frédéric; Mariette, Clara; Cahn, Jean-Yves; Meunier, Mathieu; Herault, Olivier; Fenaux, Pierre; Wagner-Ballon, Orianne; Bardet, Valerie; Dreyfus, Francois; Fontenay, Michaela Dyserythropoiesis evaluated by the RED score and hepcidin:ferritin ratio predicts response to erythropoietin in lower-risk myelodysplastic syndromes Article de journal Dans: Haematologica, vol. 104, no. 3, p. 497–504, 2019, ISSN: 1592-8721. @article{park_dyserythropoiesis_2019b,Erythropoiesis-stimulating agents are generally the first line of treatment of anemia in patients with lower-risk myelodysplastic syndrome. We prospectively investigated the predictive value of somatic mutations, and biomarkers of ineffective erythropoiesis including the flow cytometry RED score, serum growth-differentiation factor-15, and hepcidin levels. Inclusion criteria were no prior treatment with erythropoiesis-stimulating agents, low- or intermediate-1-risk myelodysplastic syndrome according to the International Prognostic Scoring System, and a hemoglobin level textless10 g/dL. Patients could be red blood cell transfusion-dependent or not and were given epoetin zeta 40 000 IU/week. Serum erythropoietin level, iron parameters, hepcidin, flow cytometry Ogata and RED scores, and growth-differentiation factor-15 levels were determined at baseline, and molecular analysis by next-generation sequencing was also conducted. Erythroid response (defined according to the International Working Group 2006 criteria) was assessed at week 12. Seventy patients, with a median age of 78 years, were included in the study. There were 22 patients with refractory cytopenia with multilineage dysplasia, 19 with refractory cytopenia with unilineage dysplasia, 14 with refractory anemia with ring sideroblasts, four with refractory anemia with excess blasts-1, six with chronic myelomonocytic leukemia, two with del5q-and three with unclassifiable myelodysplastic syndrome. According to the revised International Prognostic Scoring System, 13 had very low risk, 47 had low risk, nine intermediate risk and one had high-risk disease. Twenty patients were transfusion dependent. Forty-eight percent had an erythroid response and the median duration of the response was 26 months. At baseline, non-responders had significantly higher RED scores and lower hepcidin:ferritin ratios. In multivariate analysis, only a RED score textgreater4 (P=0.05) and a hepcidin:ferritin ratio textless9 (P=0.02) were statistically significantly associated with worse erythroid response. The median response duration was shorter in patients with growth-differentiation factor-15 textgreater2000 pg/mL and a hepcidin:ferritin ratio textless9 (P=0.0008 and P=0.01, respectively). In multivariate analysis, both variables were associated with shorter response duration. Erythroid response to epoetin zeta was similar to that obtained with other erythropoiesis-stimulating agents and was correlated with higher baseline hepcidin:ferritin ratio and lower RED score. ClinicalTrials.gov registration: NCT 03598582. |
Tebani, A.; Abily-Donval, L.; Schmitz-Afonso, I.; Piraud, M.; Ausseil, J.; Zerimech, F.; Pilon, C.; Pereira, T.; Marret, S.; Afonso, C.; Bekri, S. Analysis of Mucopolysaccharidosis Type VI through Integrative Functional Metabolomics Article de journal Dans: Int J Mol Sci, vol. 20, no. 2, 2019, ISSN: 1422-0067 (Electronic) 1422-0067 (Linking). @article{RN1000, |
Braun, H.; Geniez, C.; Degboe, Y.; Constantin, A.; Cantagrel, A.; Nigon, D.; Sans, N.; Faruch-Bilfeld, M.; Ruyssen-Witrand, A. Prevalence of inflammatory posterior arch abnormalities on lumbar spine MRI in spondyloarthritis patients compared with low back pain patients Article de journal Dans: Eur Radiol, vol. 29, no. 12, p. 6405-6415, 2019, ISSN: 1432-1084 (Electronic) 0938-7994 (Linking). @article{RN1010, |
Casassa, E. A.; Mailhol, C.; Tournier, E.; Laurent, C.; Degboe, Y.; Eischen, M.; Kirsten, N.; Moreau, J.; Evrard, S. M.; Mansat-De Mas, V.; Lamant, L.; Dubreuil, P.; Apoil, P. A.; Hermine, O.; Paul, C.; Bulai Livideanu, C. Mast cell activation syndrome: High frequency of skin manifestations and anaphylactic shock Article de journal Dans: Allergol Int, vol. 68, no. 1, p. 119-121, 2019, ISSN: 1440-1592 (Electronic) 1323-8930 (Linking). @article{RN1009b, |
Champs, B.; Degboe, Y.; Barnetche, T.; Cantagrel, A.; Ruyssen-Witrand, A.; Constantin, A. Dans: RMD Open, vol. 5, no. 1, p. e000763, 2019, ISSN: 2056-5933 (Print) 2056-5933 (Linking). @article{RN1008b, |
Degboe, Y.; Eischen, M.; Apoil, P. A.; Mailhol, C.; Dubreuil, P.; Hermine, O.; Paul, C.; Bulai Livideanu, C.; Laroche, M. Higher prevalence of vertebral fractures in systemic mastocytosis, but not in cutaneous mastocytosis and idiopathic mast cell activation syndrome Article de journal Dans: Osteoporos Int, vol. 30, no. 6, p. 1235-1241, 2019, ISSN: 1433-2965 (Electronic) 0937-941X (Linking). @article{RN1007b, |
Degboe, Y.; Rauwel, B.; Baron, M.; Boyer, J. F.; Ruyssen-Witrand, A.; Constantin, A.; Davignon, J. L. Polarization of Rheumatoid Macrophages by TNF Targeting Through an IL-10/STAT3 Mechanism Article de journal Dans: Front Immunol, vol. 10, p. 3, 2019, ISSN: 1664-3224 (Electronic) 1664-3224 (Linking). @article{RN1006b, |
Fradet, M.; Negretto, M.; Tournier, E.; Laurent, C.; Apoil, P. A.; Evrard, S.; Degboe, Y.; Del Mas, V.; Lamant, L.; Dubreuil, P.; Laroche, M.; Mailhol, C.; Hermine, O.; Paul, C.; Bulai Livideanu, C. Frequency of isolated cutaneous involvement in adult mastocytosis: a cohort study Article de journal Dans: J Eur Acad Dermatol Venereol, vol. 33, no. 9, p. 1713-1718, 2019, ISSN: 1468-3083 (Electronic) 0926-9959 (Linking). @article{RN1005, |
Luxembourger, C.; Ruyssen-Witrand, A.; Ladhari, C.; Rittore, C.; Degboe, Y.; Maillefert, J. F.; Gaudin, P.; Marotte, H.; Wendling, D.; Jorgensen, C.; Cantagrel, A.; Constantin, A.; Nigon, D.; Touitou, I.; Gottenberg, J. E.; Pers, Y. M. A single nucleotide polymorphism of IL6-receptor is associated with response to tocilizumab in rheumatoid arthritis patients Article de journal Dans: Pharmacogenomics J, vol. 19, no. 4, p. 368-374, 2019, ISSN: 1473-1150 (Electronic) 1470-269X (Linking). @article{RN1004, |
Magnol, M.; Degboe, Y.; Couture, G.; Constantin, A.; Laroche, M. Complex regional pain syndrome secondary to everolimus: Two cases Article de journal Dans: Joint Bone Spine, vol. 86, no. 5, p. 659-660, 2019, ISSN: 1778-7254 (Electronic) 1297-319X (Linking). @article{RN1003b, |
Ruyssen-Witrand, A.; Luxembourger, C.; Cantagrel, A.; Nigon, D.; Claudepierre, P.; Degboe, Y.; Constantin, A. Association between IL23R and ERAP1 polymorphisms and sacroiliac or spinal MRI inflammation in spondyloarthritis: DESIR cohort data Article de journal Dans: Arthritis Res Ther, vol. 21, no. 1, p. 22, 2019, ISSN: 1478-6362 (Electronic) 1478-6354 (Linking). @article{RN1002b, |
Sayegh, S.; El Atat, O.; Diallo, K.; Rauwel, B.; Degboe, Y.; Cavaignac, E.; Constantin, A.; Cantagrel, A.; Trak-Smayra, V.; Alaaeddine, N.; Davignon, J. L. Corrigendum: Rheumatoid Synovial Fluids Regulate the Immunomodulatory Potential of Adipose-Derived Mesenchymal Stem Cells Through a TNF/NF-kappaB-Dependent Mechanism Article de journal Dans: Front Immunol, vol. 10, p. 1961, 2019, ISSN: 1664-3224 (Electronic) 1664-3224 (Linking). @article{RN1001b, |
Sayegh, S.; El Atat, O.; Diallo, K.; Rauwel, B.; Degboe, Y.; Cavaignac, E.; Constantin, A.; Cantagrel, A.; Trak-Smayra, V.; Alaaeddine, N.; Davignon, J. L. Rheumatoid Synovial Fluids Regulate the Immunomodulatory Potential of Adipose-Derived Mesenchymal Stem Cells Through a TNF/NF-kappaB-Dependent Mechanism Article de journal Dans: Front Immunol, vol. 10, p. 1482, 2019, ISSN: 1664-3224 (Electronic) 1664-3224 (Linking). @article{RN1000b, |
Lefebvre, Thibaud; Millot, Sarah; Richard, Emmanuel; Blouin, Jean-Marc; Lalanne, Magalie; Lamrissi-Garcia, Isabelle; Costet, Pierre; Lyoumi, Said; Gouya, Laurent; Puy, Hervé; Moreau-Gaudry, François; de Verneuil, Hubert; Karim, Zoubida; Ged, Cécile Genetic background influences hepcidin response to iron imbalance in a mouse model of hemolytic anemia (Congenital erythropoietic porphyria) Article de journal Dans: Biochemical and Biophysical Research Communications, vol. 520, no. 2, p. 297–303, 2019, ISSN: 1090-2104. @article{lefebvre_genetic_2019,Clinical severity is heterogeneous among patients suffering from congenital erythropoietic porphyria (CEP) suggesting a modulation of the disease (UROS deficiency) by environmental factors and modifier genes. A KI model of CEP due to a missense mutation of UROS gene present in human has been developed on 3 congenic mouse strains (BALB/c, C57BL/6, and 129/Sv) in order to study the impact of genetic background on disease severity. To detect putative modifiers of disease expression in congenic mice, hematologic data, iron parameters, porphyrin content and tissue samples were collected. Regenerative hemolytic anemia, a consequence of porphyrin excess in RBCs, had various expressions: 129/Sv mice were more hemolytic, BALB/c had more regenerative response to anemia, C57BL/6 were less affected. Iron status and hemolysis level were directly related: C57BL/6 and BALB/c had moderate hemolysis and active erythropoiesis able to reduce iron overload in the liver, while, 129/Sv showed an imbalance between iron release due to hemolysis and erythroid use. The negative control of hepcidin on the ferroportin iron exporter appeared strain specific in the CEP mice models tested. Full repression of hepcidin was observed in BALB/c and 129/Sv mice, favoring parenchymal iron overload in the liver. Unchanged hepcidin levels in C57BL/6 resulted in retention of iron predominantly in reticuloendothelial tissues. These findings open the field for potential therapeutic applications in the human disease, of hepcidin agonists and iron depletion in chronic hemolytic anemia. |
Daher, Raêd; Lefebvre, Thibaud; Puy, Hervé; Karim, Zoubida Extrahepatic hepcidin production: The intriguing outcomes of recent years Article de journal Dans: World Journal of Clinical Cases, vol. 7, no. 15, p. 1926–1936, 2019, ISSN: 2307-8960. @article{daher_extrahepatic_2019,Hepcidin is the hyposideremic hormone regulating iron metabolism. It is a defensin-like disulfide-bonded peptide with antimicrobial activity. The main site of hepcidin production is the liver where its synthesis is modulated by iron, inflammation and erythropoietic signaling. However, hepcidin locally produced in several peripheral organs seems to be an important actor for the maintenance of iron homeostasis in these organs. This review highlights the presence of peripheral hepcidin and its potential functions. Understanding the role of extrahepatic hepcidin could be of great physiological and therapeutic importance for several specific pathologies. |
Bondu, Sabrina; Alary, Anne-Sophie; Lefèvre, Carine; Houy, Alexandre; Jung, Grace; Lefebvre, Thibaud; Rombaut, David; Boussaid, Ismael; Bousta, Abderrahmane; Guillonneau, François; Perrier, Prunelle; Alsafadi, Samar; Wassef, Michel; Margueron, Raphaël; Rousseau, Alice; Droin, Nathalie; Cagnard, Nicolas; Kaltenbach, Sophie; Winter, Susann; Kubasch, Anne-Sophie; Bouscary, Didier; Santini, Valeria; Toma, Andrea; Hunault, Mathilde; Stamatoullas, Aspasia; Gyan, Emmanuel; Cluzeau, Thomas; Platzbecker, Uwe; Adès, Lionel; Puy, Hervé; Stern, Marc-Henri; Karim, Zoubida; Mayeux, Patrick; Nemeth, Elizabeta; Park, Sophie; Ganz, Tomas; Kautz, Léon; Kosmider, Olivier; Fontenay, Michaëla A variant erythroferrone disrupts iron homeostasis in SF3B1-mutated myelodysplastic syndrome Article de journal Dans: Science Translational Medicine, vol. 11, no. 500, 2019, ISSN: 1946-6242. @article{bondu_variant_2019,Myelodysplastic syndromes (MDS) with ring sideroblasts are hematopoietic stem cell disorders with erythroid dysplasia and mutations in the SF3B1 splicing factor gene. Patients with MDS with SF3B1 mutations often accumulate excessive tissue iron, even in the absence of transfusions, but the mechanisms that are responsible for their parenchymal iron overload are unknown. Body iron content, tissue distribution, and the supply of iron for erythropoiesis are controlled by the hormone hepcidin, which is regulated by erythroblasts through secretion of the erythroid hormone erythroferrone (ERFE). Here, we identified an alternative ERFE transcript in patients with MDS with the SF3B1 mutation. Induction of this ERFE transcript in primary SF3B1-mutated bone marrow erythroblasts generated a variant protein that maintained the capacity to suppress hepcidin transcription. Plasma concentrations of ERFE were higher in patients with MDS with an SF3B1 gene mutation than in patients with SF3B1 wild-type MDS. Thus, hepcidin suppression by a variant ERFE is likely responsible for the increased iron loading in patients with SF3B1-mutated MDS, suggesting that ERFE could be targeted to prevent iron-mediated toxicity. The expression of the variant ERFE transcript that was restricted to SF3B1-mutated erythroblasts decreased in lenalidomide-responsive anemic patients, identifying variant ERFE as a specific biomarker of clonal erythropoiesis. |
Mirmiran, Arienne; Schmitt, Caroline; Lefebvre, Thibaud; Manceau, Hana; Daher, Raêd; Oustric, Vincent; Poli, Antoine; Lacapère, Jean-Jacques; Moulouel, Boualem; Puy, Hervé; Karim, Zoubida; Peoc'h, Katell; Lenglet, Hugo; Simonin, Sylvie; Deybach, Jean-Charles; Nicolas, Gaël; Gouya, Laurent Erythroid-Progenitor-Targeted Gene Therapy Using Bifunctional TFR1 Ligand-Peptides in Human Erythropoietic Protoporphyria Article de journal Dans: American Journal of Human Genetics, vol. 104, no. 2, p. 341–347, 2019, ISSN: 1537-6605. @article{mirmiran_erythroid-progenitor-targeted_2019,Erythropoietic protoporphyria (EPP) is a hereditary disease characterized by a deficiency in ferrochelatase (FECH) activity. FECH activity is responsible for the accumulation of protoporphyrin IX (PPIX). Without etiopathogenic treatment, EPP manifests as severe photosensitivity. 95% of affected individuals present a hypomorphic FECH allele trans to a loss-of-function (LOF) FECH mutation, resulting in a reduction in FECH activity in erythroblasts below a critical threshold. The hypomorphic allele promotes the use of a cryptic acceptor splice site, generating an aberrant FECH mRNA, which is responsible for the reduced level of wild-type FECH mRNA and, ultimately, FECH activity. We have previously identified an antisense oligonucleotide (AON), AON-V1 (V1), that redirects splicing to the physiological acceptor site and reduces the accumulation of PPIX. Here, we developed a specific strategy that uses transferrin receptor 1 (TRF1) as a Trojan horse to deliver V1 to erythroid progenitors. We designed a bifunctional peptide (P1-9R) including a TFR1-targeting peptide coupled to a nine-arginine cell-penetrating peptide (CPP) that facilitates the release of the AON from TFR1 in endosomal vesicles. We demonstrated that the P1-9R/V1 nanocomplex promotes the efficient and prolonged redirection of splicing towards the physiological splice site and subsequent normalization of WT FECH mRNA and protein levels. Finally, the P1-9R/V1 nanocomplex increases WT FECH mRNA production and significantly decreases PPIX accumulation in primary cultures of differentiating erythroid progenitors from an overt EPP-affected individual. P1-9R is a method designed to target erythroid progenitors and represents a potentially powerful tool for the in vivo delivery of therapeutic DNA in many erythroid disorders. |
Rio, Sarah; Gastou, Marc; Karboul, Narjesse; Derman, Raphaёl; Suriyun, Thunwarat; Manceau, Hana; Leblanc, Thierry; El Benna, Jamel; Schmitt, Caroline; Azouzi, Slim; Larghéro, Jérome; Karim, Zoubida; Macias-Garcia, Alejandra; Chen, Jane-Jane; Hermine, Olivier; Courtois, Geneviève; Puy, Hervé; Gouya, Laurent; Mohandas, Narla; Da Costa, Lydie Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1 Article de journal Dans: Blood, vol. 133, no. 12, p. 1358–1370, 2019, ISSN: 1528-0020. @article{rio_regulation_2019,Diamond-Blackfan anemia (DBA) is a congenital erythroblastopenia that is characterized by a blockade in erythroid differentiation related to impaired ribosome biogenesis. DBA phenotype and genotype are highly heterogeneous. We have previously identified 2 in vitro erythroid cell growth phenotypes for primary CD34+ cells from DBA patients and following short hairpin RNA knockdown of RPS19, RPL5, and RPL11 expression in normal human CD34+ cells. The haploinsufficient RPS19 in vitro phenotype is less severe than that of 2 other ribosomal protein (RP) mutant genes. We further documented that proteasomal degradation of HSP70, the chaperone of GATA1, is a major contributor to the defect in erythroid proliferation, delayed erythroid differentiation, increased apoptosis, and decreased globin expression, which are all features of the RPL5 or RPL11 DBA phenotype. In the present study, we explored the hypothesis that an imbalance between globin and heme synthesis may be involved in pure red cell aplasia of DBA. We identified disequilibrium between the globin chain and the heme synthesis in erythroid cells of DBA patients. This imbalance led to accumulation of excess free heme and increased reactive oxygen species production that was more pronounced in cells of the RPL5 or RPL11 phenotype. Strikingly, rescue experiments with wild-type HSP70 restored GATA1 expression levels, increased globin synthesis thereby reducing free heme excess and resulting in decreased apoptosis of DBA erythroid cells. These results demonstrate the involvement of heme in DBA pathophysiology and a major role of HSP70 in the control of balanced heme/globin synthesis. |
Lefebvre, Thibaud; Millot, Sarah; Richard, Emmanuel; Blouin, Jean-Marc; Lalanne, Magalie; Lamrissi-Garcia, Isabelle; Costet, Pierre; Lyoumi, Said; Gouya, Laurent; Puy, Hervé; Moreau-Gaudry, François; de Verneuil, Hubert; Karim, Zoubida; Ged, Cécile Genetic background influences hepcidin response to iron imbalance in a mouse model of hemolytic anemia (Congenital erythropoietic porphyria) Article de journal Dans: Biochemical and Biophysical Research Communications, vol. 520, no. 2, p. 297–303, 2019, ISSN: 1090-2104. @article{lefebvre_genetic_2019b,Clinical severity is heterogeneous among patients suffering from congenital erythropoietic porphyria (CEP) suggesting a modulation of the disease (UROS deficiency) by environmental factors and modifier genes. A KI model of CEP due to a missense mutation of UROS gene present in human has been developed on 3 congenic mouse strains (BALB/c, C57BL/6, and 129/Sv) in order to study the impact of genetic background on disease severity. To detect putative modifiers of disease expression in congenic mice, hematologic data, iron parameters, porphyrin content and tissue samples were collected. Regenerative hemolytic anemia, a consequence of porphyrin excess in RBCs, had various expressions: 129/Sv mice were more hemolytic, BALB/c had more regenerative response to anemia, C57BL/6 were less affected. Iron status and hemolysis level were directly related: C57BL/6 and BALB/c had moderate hemolysis and active erythropoiesis able to reduce iron overload in the liver, while, 129/Sv showed an imbalance between iron release due to hemolysis and erythroid use. The negative control of hepcidin on the ferroportin iron exporter appeared strain specific in the CEP mice models tested. Full repression of hepcidin was observed in BALB/c and 129/Sv mice, favoring parenchymal iron overload in the liver. Unchanged hepcidin levels in C57BL/6 resulted in retention of iron predominantly in reticuloendothelial tissues. These findings open the field for potential therapeutic applications in the human disease, of hepcidin agonists and iron depletion in chronic hemolytic anemia. |
Daher, Raêd; Lefebvre, Thibaud; Puy, Hervé; Karim, Zoubida Extrahepatic hepcidin production: The intriguing outcomes of recent years Article de journal Dans: World Journal of Clinical Cases, vol. 7, no. 15, p. 1926–1936, 2019, ISSN: 2307-8960. @article{daher_extrahepatic_2019b,Hepcidin is the hyposideremic hormone regulating iron metabolism. It is a defensin-like disulfide-bonded peptide with antimicrobial activity. The main site of hepcidin production is the liver where its synthesis is modulated by iron, inflammation and erythropoietic signaling. However, hepcidin locally produced in several peripheral organs seems to be an important actor for the maintenance of iron homeostasis in these organs. This review highlights the presence of peripheral hepcidin and its potential functions. Understanding the role of extrahepatic hepcidin could be of great physiological and therapeutic importance for several specific pathologies. |
Touzot, Maxime; Lefebvre, Thibaud; Roux, Arthur; Maheas, Catherine; Ridel, Christophe; Puy, Hervé; Karim, Zoubida Functional erythropoietin-hepcidin axis in recombinant human erythropoietin independent haemodialysis patients Article de journal Dans: Nephrology (Carlton, Vic.), vol. 24, no. 7, p. 751–757, 2019, ISSN: 1440-1797. @article{touzot_functional_2019b,AIM: Relatively few haemodialysis (HD) patients remain independent of recombinant human erythropoietin ('rHU-EPO free patients'). We investigated the role of EPO and hepcidin, two key hormones involved in anaemia. METHODS: We report a monocentric case-control series. Iron status, EPO and hepcidin levels were analysed in 15 Adult HD (Age textgreater 18 years) with a stable haemoglobin (Hb) level that have not received rHU-EPO for at least 6 months (=rHU-EPO free patients); and in 60 controls with a stable rHU-EPO dose and Hb level. RESULTS: The rHU-EPO free patients had a higher Hb level compared to controls (12.1 ± 0.99 g/dL vs 11.1 ± 0.73, P = 0.0014), and a lower ferritin level (183 ± 102 vs 312 ± 166 ng/mL, P = 0.001). Hepcidin levels were lower in the rHU-EPO free patients (12.53 ± 10.46 ng/mL) compared to the controls (37.95 ± 34.33 ng/mL), P = 0.0033. Hepcidin levels correlated significantly with ferritin levels; but neither with transferrin saturation, C-reactive protein nor EPO levels. Unsupervised analysis revealed that rHU-EPO free patients had a specific clinical/biological profile (presence of renal cyst, longer dialysis vintage, lower ferritin, and EPO and hepcidin levels compared to the control group). Finally, we showed that a lower ferritin level might be a surrogate marker of a lower hepcidin status in this population. CONCLUSION: Recombinant human erythropoietin free patients seem to restore the EPO-hepcidin axis that is critical for erythropoiesis. A specific combination of clinical and biological parameters may help to detect future rHU-EPO free patients. |
Mirmiran, Arienne; Schmitt, Caroline; Lefebvre, Thibaud; Manceau, Hana; Daher, Raêd; Oustric, Vincent; Poli, Antoine; Lacapère, Jean-Jacques; Moulouel, Boualem; Puy, Hervé; Karim, Zoubida; Peoc'h, Katell; Lenglet, Hugo; Simonin, Sylvie; Deybach, Jean-Charles; Nicolas, Gaël; Gouya, Laurent Erythroid-Progenitor-Targeted Gene Therapy Using Bifunctional TFR1 Ligand-Peptides in Human Erythropoietic Protoporphyria Article de journal Dans: American Journal of Human Genetics, vol. 104, no. 2, p. 341–347, 2019, ISSN: 1537-6605. @article{mirmiran_erythroid-progenitor-targeted_2019b,Erythropoietic protoporphyria (EPP) is a hereditary disease characterized by a deficiency in ferrochelatase (FECH) activity. FECH activity is responsible for the accumulation of protoporphyrin IX (PPIX). Without etiopathogenic treatment, EPP manifests as severe photosensitivity. 95% of affected individuals present a hypomorphic FECH allele trans to a loss-of-function (LOF) FECH mutation, resulting in a reduction in FECH activity in erythroblasts below a critical threshold. The hypomorphic allele promotes the use of a cryptic acceptor splice site, generating an aberrant FECH mRNA, which is responsible for the reduced level of wild-type FECH mRNA and, ultimately, FECH activity. We have previously identified an antisense oligonucleotide (AON), AON-V1 (V1), that redirects splicing to the physiological acceptor site and reduces the accumulation of PPIX. Here, we developed a specific strategy that uses transferrin receptor 1 (TRF1) as a Trojan horse to deliver V1 to erythroid progenitors. We designed a bifunctional peptide (P1-9R) including a TFR1-targeting peptide coupled to a nine-arginine cell-penetrating peptide (CPP) that facilitates the release of the AON from TFR1 in endosomal vesicles. We demonstrated that the P1-9R/V1 nanocomplex promotes the efficient and prolonged redirection of splicing towards the physiological splice site and subsequent normalization of WT FECH mRNA and protein levels. Finally, the P1-9R/V1 nanocomplex increases WT FECH mRNA production and significantly decreases PPIX accumulation in primary cultures of differentiating erythroid progenitors from an overt EPP-affected individual. P1-9R is a method designed to target erythroid progenitors and represents a potentially powerful tool for the in vivo delivery of therapeutic DNA in many erythroid disorders. |
Rio, Sarah; Gastou, Marc; Karboul, Narjesse; Derman, Raphaёl; Suriyun, Thunwarat; Manceau, Hana; Leblanc, Thierry; El Benna, Jamel; Schmitt, Caroline; Azouzi, Slim; Larghéro, Jérome; Karim, Zoubida; Macias-Garcia, Alejandra; Chen, Jane-Jane; Hermine, Olivier; Courtois, Geneviève; Puy, Hervé; Gouya, Laurent; Mohandas, Narla; Da Costa, Lydie Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1 Article de journal Dans: Blood, vol. 133, no. 12, p. 1358–1370, 2019, ISSN: 1528-0020. @article{rio_regulation_2019b,Diamond-Blackfan anemia (DBA) is a congenital erythroblastopenia that is characterized by a blockade in erythroid differentiation related to impaired ribosome biogenesis. DBA phenotype and genotype are highly heterogeneous. We have previously identified 2 in vitro erythroid cell growth phenotypes for primary CD34+ cells from DBA patients and following short hairpin RNA knockdown of RPS19, RPL5, and RPL11 expression in normal human CD34+ cells. The haploinsufficient RPS19 in vitro phenotype is less severe than that of 2 other ribosomal protein (RP) mutant genes. We further documented that proteasomal degradation of HSP70, the chaperone of GATA1, is a major contributor to the defect in erythroid proliferation, delayed erythroid differentiation, increased apoptosis, and decreased globin expression, which are all features of the RPL5 or RPL11 DBA phenotype. In the present study, we explored the hypothesis that an imbalance between globin and heme synthesis may be involved in pure red cell aplasia of DBA. We identified disequilibrium between the globin chain and the heme synthesis in erythroid cells of DBA patients. This imbalance led to accumulation of excess free heme and increased reactive oxygen species production that was more pronounced in cells of the RPL5 or RPL11 phenotype. Strikingly, rescue experiments with wild-type HSP70 restored GATA1 expression levels, increased globin synthesis thereby reducing free heme excess and resulting in decreased apoptosis of DBA erythroid cells. These results demonstrate the involvement of heme in DBA pathophysiology and a major role of HSP70 in the control of balanced heme/globin synthesis. |
Sayegh, S.; Atat, O. El; Diallo, K.; Rauwel, B.; Degboe, Y.; Cavaignac, E.; Constantin, A.; Cantagrel, A.; Trak-Smayra, V.; Alaaeddine, N.; Davignon, J. L. Rheumatoid Synovial Fluids Regulate the Immunomodulatory Potential of Adipose-Derived Mesenchymal Stem Cells Through a TNF/NF-kappaB-Dependent Mechanism Article de journal Dans: Front Immunol, vol. 10, p. 1482, 2019, ISSN: 1664-3224 (Electronic) 1664-3224 (Linking), (Sayegh, Souraya El Atat, Oula Diallo, Katy Rauwel, Benjamin Degboe, Yannick Cavaignac, Etienne Constantin, Arnaud Cantagrel, Alain Trak-Smayra, Viviane Alaaeddine, Nada Davignon, Jean-Luc eng Research Support, Non-U.S. Gov't Switzerland 2019/07/19 Front Immunol. 2019 Jun 28;10:1482. doi: 10.3389/fimmu.2019.01482. eCollection 2019.). @article{RN2132,Introduction: Adipose-derived mesenchymal stem cells (ADSC) have been shown to have remarkable immune-modulating effects. However, their efficacy in clinical trials has yet to be fully demonstrated. This could be due to a lack of a proper inflammatory environment in vivo that primes ADSC. Here, we define how the articular microenvironment of rheumatoid arthritis (RA) patients modulates the therapeutic efficiency of ADSC. Methods: Synovial fluids (SF) were collected from 8 RA patients, 2 Spondyloarthritis patients and one control synovial fluid from a patient undergoing traumatic-related surgery. SF inflammatory status was determined by routine analysis and quantification of pro-inflammatory cytokines. ADSC were first treated with SF and ADSC proliferation and gene expression of immunomodulatory factors was evaluated. In order to determine the mechanisms underlying the effect of SF on ADSC, tumor necrosis factor (TNF), interleukin-6 (IL-6), and NF-kappaB neutralization assays were performed. To evaluate the effect of SF on ADSC functions, ADSC were pre-treated with SF and then co-cultured with either macrophages or T cells. The modulation of their phenotype was assessed by flow cytometry. Results: Pro-inflammatory RASF maintained the proliferative capacity of ADSC and upregulated the gene expression of cyclooxygenase-2 (COX2), indoleamine-1,2-dioxygenase (IDO), interleukin-6 (IL-6), tumor-necrosis factor stimulated gene 6 (TSG6), intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and programmed death-ligand 1 (PD-L1), all factors involved in ADSC immunomodulatory potential. The RASF-induced gene expression was mainly mediated by TNF alone or in combination with IL-6 and signaled through the NF-kappaB pathway. Conditioning ADSC with pro-inflammatory RASF enhanced their ability to induce CD4(+)Foxp3(+)CD25(high) regulatory T cells (Tregs) and inhibit pro-inflammatory markers CD40 and CD80 in activated macrophages. Conclusions: Inflammatory synovial fluids from RA patients had the capacity to modulate ADSC response, to induce Tregs and modulate the phenotype of macrophages. The clinical use of ADSC in affected joints should take into account the influence of the local articular environment on their potential. Having a sufficient pro-inflammatory microenvironment will determine whether optimal immunoregulatory response should be expected. Direct ADSC intra-articular delivery to patients could be a potential strategy to properly prime their immunomodulatory potential and enhance their clinical benefits. |
Luxembourger, C.; Ruyssen-Witrand, A.; Ladhari, C.; Rittore, C.; Degboe, Y.; Maillefert, J. F.; Gaudin, P.; Marotte, H.; Wendling, D.; Jorgensen, C.; Cantagrel, A.; Constantin, A.; Nigon, D.; Touitou, I.; Gottenberg, J. E.; Pers, Y. M. A single nucleotide polymorphism of IL6-receptor is associated with response to tocilizumab in rheumatoid arthritis patients Article de journal Dans: Pharmacogenomics J, vol. 19, no. 4, p. 368-374, 2019, ISSN: 1473-1150 (Electronic) 1470-269X (Linking), (Luxembourger, Cecile Ruyssen-Witrand, Adeline Ladhari, Chayma Rittore, Cecile Degboe, Yannick Maillefert, Jean-Francis Gaudin, Philippe Marotte, Hubert Wendling, Daniel Jorgensen, Christian Cantagrel, Alain Constantin, Arnaud Nigon, Delphine Touitou, Isabelle Gottenberg, Jacques-Eric Pers, Yves-Marie eng Meta-Analysis 2019/01/17 Pharmacogenomics J. 2019 Aug;19(4):368-374. doi: 10.1038/s41397-019-0072-6. Epub 2019 Jan 16.). @article{RN2134,Biological disease-modifying anti-rheumatic drugs (bDMARDs) have changed care of patients with rheumatoid arthritis (RA). However, bDMARDs are costly, can lead to serious infections, and induce a sustained remission in only 30% of RA patients. In this study, we sought to determine if the clinical response to treatment with Tocilizumab (TCZ), an IL-6 inhibitor, varied with genetic background. The efficacy of TCZ was assessed using the European League Against Rheumatism (EULAR) response criteria, measured after 3 months of treatment in two samples of French RA patients (TOCI and ROC studies). Single nucleotide polymorphisms (SNPs) in 21 candidate genes were genotyped using KasPar method (LGC-genomics, UK) and then analyzed to determine their contribution to variation in the response to treatment. One hundred twenty-three patients in the TOCI group (79.8%) and 48 patients in the ROC group (80%) experienced good or moderate EULAR response. The clinical response to treatment was associated with SNP genotype in the gene IL6R, with patients with the homozygous AA-genotype for rs12083537 (IL6R) showing a significantly better response than homozygous or heterozygous patients with the G allele [TOCI: 87.5% of responders for AA genotype vs. 72.2% for AG or GG genotype (p = 0.018); ROC patients: 89.2% of responders for AA genotype vs. 65.2% for AG or GG genotype |
Degboe, Y.; Eischen, M.; Apoil, P. A.; Mailhol, C.; Dubreuil, P.; Hermine, O.; Paul, C.; Livideanu, C. Bulai; Laroche, M. Higher prevalence of vertebral fractures in systemic mastocytosis, but not in cutaneous mastocytosis and idiopathic mast cell activation syndrome Article de journal Dans: Osteoporos Int, vol. 30, no. 6, p. 1235-1241, 2019, ISSN: 1433-2965 (Electronic) 0937-941X (Linking), (Degboe, Y Eischen, M Apoil, P A Mailhol, C Dubreuil, P Hermine, O Paul, C Bulai Livideanu, C Laroche, M eng England 2019/03/09 Osteoporos Int. 2019 Jun;30(6):1235-1241. doi: 10.1007/s00198-019-04918-7. Epub 2019 Mar 7.). @article{RN2136,Little is known about osteoporosis in mast cell disorders (MCDs) not related to systemic mastocytosis. We described osteoporosis and fractures in MCDs and showed that systemic mastocytosis was the only studied MCDs associated with osteoporotic vertebral fractures. INTRODUCTION: To describe osteoporosis (OP) and fragility fractures in mast cell disorders (MCDs). METHODS: We retrospectively analyzed data concerning all successive patients with systemic mastocytosis (SM), cutaneous mastocytosis (CM), and mast cell activation syndromes (MCAS) diagnosed in our mastocytosis expert center between 2004 and 2015. We collected data concerning demographic profiles, clinical signs of MCD, osteoporosis, fractures, densitometry, and biological assessment of MCD. We compared CM and MCAS patients with SM patients with regard to the characteristics of OP and fragility fractures. RESULTS: We assessed 89 SM patients, 20 CM patients, and 20 MCAS patients. Osteoporosis was less frequent in CM (15.0%) and MCAS (10.0%) than in SM (44.9%). Similarly, fractures were less frequent in non-SM MCDs, respectively 5.0%, 5.0%, and 28.1%. SM patients displayed high prevalence of vertebral fractures (22.5%), mostly multiple. Conversely, in non-SM patients, vertebral fractures appeared to be uncommon (5%) and more frequently associated with risk factors for osteoporosis. CONCLUSIONS: SM is associated with multiple vertebral osteoporotic fractures, whereas CM and MCAS do not appear to be associated with this phenotype. |
Degboe, Y.; Rauwel, B.; Baron, M.; Boyer, J. F.; Ruyssen-Witrand, A.; Constantin, A.; Davignon, J. L. Polarization of Rheumatoid Macrophages by TNF Targeting Through an IL-10/STAT3 Mechanism Article de journal Dans: Front Immunol, vol. 10, p. 3, 2019, ISSN: 1664-3224 (Electronic) 1664-3224 (Linking), (Degboe, Yannick Rauwel, Benjamin Baron, Michel Boyer, Jean-Frederic Ruyssen-Witrand, Adeline Constantin, Arnaud Davignon, Jean-Luc eng Research Support, Non-U.S. Gov't Switzerland 2019/02/05 Front Immunol. 2019 Jan 18;10:3. doi: 10.3389/fimmu.2019.00003. eCollection 2019.). @article{RN2135,Macrophages contribute to the pathogenesis of rheumatoid arthritis (RA). They can display different states of activation or "polarization," notably the so-called inflammatory "M1" and the various alternative "M2" polarizations, characterized by distinct functions. Data regarding the effects of RA anti-cytokine biological disease-modifying anti-rheumatic drugs (bDMARDs) on macrophage polarization are scarce. We aimed to assess in vitro modulation of macrophage polarization by bDMARDs targeting pro-inflammatory cytokines in RA. We generated monocyte derived macrophages using blood samples from 20 RA patients with active RA and 30 healthy controls. We evaluated in vitro the impact on M1 inflammatory macrophages of: etanercept (ETA), adalimumab (ADA), certolizumab (CZP), tocilizumab (TCZ), and rituximab (RTX). We assessed the impact on macrophage polarization using flow cytometry and RTqPCR to study the expression of surface markers and perform functional studies of cytokine production, phagocytosis, and negative feedback control of inflammation. Among evaluated bDMARDs, anti-TNF agents modulated the polarization of inflammatory macrophages by decreasing inflammatory surface markers (CD40, CD80) and favoring alternative markers (CD16, CD163, MerTK). Anti-TNF agents also induced alternative functions in macrophages activated in inflammatory condition with (i) the inhibition of inflammatory cytokines (TNF, IL-6, IL-12), (ii) an increase in phagocytosis. These findings were mechanistically related to an increase in early IL-10 production, responsible for higher negative feedback control of inflammation involving SOCS3 and Gas6. This IL-10 effect was STAT3-dependent. Anti-TNF agents not only inhibit in vitro inflammatory functions of macrophages, but also favor resolution of inflammation through polarization toward alternative features specifically involving the IL-10/STAT3 axis. |
2018 |
Moniez, Sophie; Pienkowski, Catherine; Lepage, Benoit; Hamdi, Safouane; Daudin, Myriam; Oliver, Isabelle; Jouret, Béatrice; Cartault, Audrey; Diene, Gwenaelle; Verloes, Alain; Cavé, Hélène; Salles, Jean-Pierre; Tauber, Maithé; Yart, Armelle; Edouard, Thomas Noonan syndrome males display Sertoli cell-specific primary testicular insufficiency Article de journal Dans: European Journal of Endocrinology, vol. 179, no. 6, p. 409–418, 2018. @article{Moniez2018,Context Abnormalities in the hypothalamo-pituitary-gonadal axis have long been reported in Noonan syndrome (NS) males with only few data available in prepubertal children. Objective The aim of this study was to describe the gonadal function of NS males from childhood to adulthood. Design It is a retrospective chart review. Patients and methods A total of 37 males with a genetically confirmed diagnosis of NS were included. Clinical and genetic features, as well as serum hormone levels (LH, FSH, testosterone, anti-Müllerian hormone (AMH), and inhibin B) were analysed. Results Of the 37 patients, 16 (43%) children had entered puberty at a median age of 13.5 years (range: 11.4-15.0 years); age at pubertal onset was negatively correlated with BMI SDS (r = -0.541; P = 0.022). In pubertal boys, testosterone levels were normal suggesting a normal Leydig cell function. In contrast, NS patients had significant lower levels of AMH (mean SDS: -0.6 ± 1.1; P = 0.003) and inhibin B (mean SDS: -1.1 ± 1.2; P textless 0.001) compared with the general population, suggesting a Sertoli cell dysfunction. Lower AMH and inhibin B levels were found in NS-PTPN11 patients, whereas these markers did not differ from healthy children in SOS1 patients. No difference was found between cryptorchid and non-cryptorchid patients for AMH and inhibin B levels (P = 0.43 and 0.62 respectively). Four NS-PTPN11 patients had a severe primary hypogonadism with azoospermia/cryptozoospermia. Conclusions NS males display Sertoli cell-specific primary testicular insufficiency, whereas Leydig cell function seems to be unaffected. |
Poupot, Rémy; Goursat, Cécile; Fruchon, Séverine Multivalent nanosystems: targeting monocytes/macrophages. Article de journal Dans: International journal of nanomedicine, vol. 13, p. 5511–5521, 2018, ISSN: 1178-2013. @article{Poupot2018,Among all the cellular partners involved in inflammatory processes, monocytes and macrophages are the master regulators of inflammation. They are found in almost all the tissues and are nearly the only cells capable of performing each step of inflammation. Consequently, they stand as major relevant therapeutic targets to treat inflammatory disorders and diseases. The physiological phagocytic activity of macrophages prompts them to detect, to recognize, and eventually to engulf any nanosystem cruising in their neighborhood. Interestingly, nanosystems can be rationally engineered to afford multivalent, and multifunctional if needed, entities with multiplexed and/or reinforced biological activities. Indeed, engineered nanosystems bearing moieties specifically targeting macrophages, and loaded with or bound to drugs are promising candidates to modulate, or even eradicate, deleterious macrophages in vivo. In this review we highlight recent articles and concepts of multivalent nanosystems targeting monocytes and macrophages to treat inflammatory disorders. |
Tauber, Maïthé; Diene, Gwenaelle; Molinas, Catherine Growth Hormone Treatment for Prader-Willi Syndrome. Article de journal Dans: Pediatric endocrinology reviews : PER, vol. 16, no. Suppl 1, p. 91–99, 2018, ISSN: 1565-4753. @article{Tauber2018,The European Marketing Authorization for recombinant human growth hormone (rhGH) in children with Prader-Willi syndrome was the first indication for metabolic and body composition effects in children. In the US it is indicated for short stature associated with PWS. Recombinant hGH is the first treatment for the PWS population and radically changed the care of these children by facilitating access to physicians who prescribe rhGH, mainly paediatric endocrinologists, and manage the organization of multidisciplinary care. Recombinant hGH treatment improved linear growth, body composition, and socialization not only in children but also in young adults. The pathophysiology of combined hormonal deficiencies including GH is starting to be unravelled. We now have to focus on co-morbidities that are not modified by rhGH treatment, such as feeding disorders and behaviour problems, to truly change the life of patients. The transition of care from adolescents to young adults also remains a challenge. |
Puy, Vincent; Darwiche, Walaa; Trudel, Stéphanie; Gomila, Cathy; Lony, Christelle; Puy, Laurent; Lefebvre, Thibaud; Vitry, Sandrine; Boullier, Agnès; Karim, Zoubida; Ausseil, Jérôme Predominant role of microglia in brain iron retention in Sanfilippo syndrome, a pediatric neurodegenerative disease Article de journal Dans: Glia, vol. 66, no. 8, p. 1709–1723, 2018, ISSN: 1098-1136. @article{puy_predominant_2018,Neuroinflammation and iron accumulation are hallmarks of a variety of adult neurodegenerative diseases. In Sanfilippo syndrome (mucopolysaccharidosis type III, MPSIII, a pediatric neurodegenerative disease that shares some features with adult neurodegenerative diseases), the progressive accumulation of heparan sulfate oligosaccharides (HSOs) induces microglia and astrocytes to produce pro-inflammatory cytokines leading to severe neuroinflammation. The objectives of the present study were (1) to measure the local iron concentration and to assess iron metabolism in the brain of a MPSIIIB murine model and (2) to identify the brain cells involved in this accumulation. We found that iron accumulation in MPSIIIB mice primarily affected the cerebral cortex where hepcidin levels were higher than in wild-type mice, and increased with aging. This increase was correlated with low expression of ferroportin 1 (FPN1), and thus brain iron retention. Moreover, we showed in vitro that HSOs are directly responsible for the production of hepcidin and the relative decrease in FPN1 expression when added to cultures of microglia and, to a lesser extent, to cultures of astrocytes. In contrast, no significant differences were observed in neurons. Hepcidin induction results from activation of the TLR4 pathway and STAT3 signaling, and leads to iron retention within microglia. Our results show that microglia have a key role in cerebral hepcidin overexpression and thus in the brain iron accumulation observed in the MPSIIIB model. |
Tajan, Mylène; Pernin-Grandjean, Julie; Beton, Nicolas; Gennero, Isabelle; Capilla, Florence; Neel, Benjamin G; Araki, Toshiyuki; Valet, Philippe; Tauber, Maithé; Salles, Jean-Pierre; Yart, Armelle; Edouard, Thomas Noonan syndrome-causing SHP2 mutants impair ERK-dependent chondrocyte differentiation during endochondral bone growth Article de journal Dans: Human Molecular Genetics, vol. 27, no. 13, p. 2276–2289, 2018. @article{Tajan2018,Growth retardation is a constant feature of Noonan syndrome (NS) but its physiopathology remains poorly understood. We previously reported that hyperactive NS-causing SHP2 mutants impair the systemic production of insulin-like growth factor 1 (IGF1) through hyperactivation of the RAS/extracellular signal-regulated kinases (ERK) signalling pathway. Besides endocrine defects, a direct effect of these mutants on growth plate has not been explored, although recent studies have revealed an important physiological role for SHP2 in endochondral bone growth. We demonstrated that growth plate length was reduced in NS mice, mostly due to a shortening of the hypertrophic zone and to a lesser extent of the proliferating zone. These histological features were correlated with decreased expression of early chondrocyte differentiation markers, and with reduced alkaline phosphatase staining and activity, in NS murine primary chondrocytes. Although IGF1 treatment improved growth of NS mice, it did not fully reverse growth plate abnormalities, notably the decreased hypertrophic zone. In contrast, we documented a role of RAS/ERK hyperactivation at the growth plate level since 1) NS-causing SHP2 mutants enhance RAS/ERK activation in chondrocytes in vivo (NS mice) and in vitro (ATDC5 cells) and 2) inhibition of RAS/ERK hyperactivation by U0126 treatment alleviated growth plate abnormalities and enhanced chondrocyte differentiation. Similar effects were obtained by chronic treatment of NS mice with statins. In conclusion, we demonstrated that hyperactive NS-causing SHP2 mutants impair chondrocyte differentiation during endochondral bone growth through a local hyperactivation of the RAS/ERK signalling pathway, and that statin treatment may be a possible therapeutic approach in NS. |
Rouillon, Jérémy; Lefebvre, Thibaud; Denard, Jérôme; Puy, Vincent; Daher, Raed; Ausseil, Jérôme; Zocevic, Aleksandar; Fogel, Paul; Peoc'h, Katell; Wong, Brenda; Servais, Laurent; Voit, Thomas; Puy, Herve; Karim, Zoubida; Svinartchouk, Fedor High urinary ferritin reflects myoglobin iron evacuation in DMD patients Article de journal Dans: Neuromuscular disorders: NMD, vol. 28, no. 7, p. 564–571, 2018, ISSN: 1873-2364. @article{rouillon_high_2018,Duchenne muscular dystrophy (DMD) is an X-linked disease caused by mutations in the dystrophin gene leading to the absence of the normal dystrophin protein. The efforts of many laboratories brought new treatments of DMD to the reality, but ongoing and forthcoming clinical trials suffer from absence of valuable biomarkers permitting to follow the outcome of the treatment day by day and to adjust the treatment if needed. In the present study the levels of 128 urinary proteins including growth factors, cytokines and chemokines were compared in urine of DMD patients and age related control subjects by antibody array approach. Surprisingly, statistically significant difference was observed only for urinary ferritin whose level was 50 times higher in young DMD patients. To explain the observed high urinary ferritin content we analysed the levels of iron, iron containing proteins and proteins involved in regulation of iron metabolism in serum and urine of DMD patients and their age-matched healthy controls. Obtained data strongly suggest that elevated level of urinary ferritin is functionally linked to the renal management of myoglobin iron derived from leaky muscles of DMD patients. This first observation of the high level of ferritin in urine of DMD patients permits to consider this protein as a new urinary biomarker in muscular dystrophies and sheds light on the mechanisms of iron metabolism and kidney functioning in DMD. |
Fruchon, Séverine; Poupot, Rémy The ABP Dendrimer, a Drug-Candidate against Inflammatory Diseases That Triggers the Activation of Interleukin-10 Producing Immune Cells. Article de journal Dans: Molecules (Basel, Switzerland), vol. 23, no. 6, 2018. @article{Fruchon2018,The ABP dendrimer, which is built on a phosphorus-based scaffold and bears twelve azabisphosphonate groups at its surface, is one of the dendrimers that has been shown to display immuno-modulatory and anti-inflammatory effects towards the human immune system. Its anti-inflammatory properties have been successfully challenged in animal models of inflammatory disorders. In this review, we trace the discovery and the evaluation of the therapeutic effects of the ABP dendrimer in three different animal models of both acute and chronic inflammatory diseases. We emphasize that its therapeutic effects rely on the enhancement of the production of Interleukin-10, the paradigm of anti-inflammatory cytokines, by different subsets of immune cells, such as monocytes/macrophages and CD4+ T lymphocytes. |
Paepegaey, A C; Coupaye, M; Jaziri, A; Ménesguen, F; Dubern, B; Polak, M; Oppert, J M; Tauber, M; Pinto, G; Poitou, C Impact of transitional care on endocrine and anthropometric parameters in Prader–Willi syndrome Article de journal Dans: Endocrine Connections, vol. 7, no. 5, p. 663–672, 2018. @article{Paepegaey2018,CONTEXT The transition of patients with Prader-Willi syndrome (PWS) to adult life for medical care is challenging because of multiple comorbidities, including hormone deficiencies, obesity and cognitive and behavioral disabilities. OBJECTIVE To assess endocrine management, and metabolic and anthropometric parameters of PWS adults who received (n = 31) or not (n = 64) transitional care, defined as specialized pediatric care followed by a structured care pathway to a multidisciplinary adult team. PATIENTS AND STUDY DESIGN Hormonal and metabolic parameters were retrospectively recorded in 95 adults with PWS (mean ± s.d. age 24.7 ± 8.2 years, BMI: 39.8 ± 12.1 kg/m²) referred to our Reference Center and compared according to transition. RESULTS Among the entire cohort, 35.8% received growth hormone (GH) during childhood and 16.8% had a GH stimulation test after completion of growth. In adulthood, 14.7% were treated with GH, 56.8% received sex-hormone therapy, whereas 91.1% were hypogonadic and 37.9% had undergone valid screening of the corticotropic axis. The main reason for suboptimal endocrine management was marked behavioral disorders. Patients receiving transitional care were more likely to have had a GH stimulation test and hormonal substitutions in childhood. They also had a lower BMI, percentage of fat mass, improved metabolic parameters and fewer antidepressant treatments. Transitional care remained significantly associated with these parameters in multivariate analysis when adjusted on GH treatment. CONCLUSION A coordinated care pathway with specialized pediatric care and transition to a multidisciplinary adult team accustomed to managing complex disability including psychiatric troubles are associated with a better health status in adults with PWS. |
Lenglet, Hugo; Schmitt, Caroline; Grange, Thomas; Manceau, Hana; Karboul, Narjesse; Bouchet-Crivat, Florian; Robreau, Anne-Marie; Nicolas, Gael; Lamoril, Jerôme; Simonin, Sylvie; Mirmiran, Arienne; Karim, Zoubida; Casalino, Enrique; Deybach, Jean-Charles; Puy, Hervé; Peoc'h, Katell; Gouya, Laurent From a dominant to an oligogenic model of inheritance with environmental modifiers in acute intermittent porphyria Article de journal Dans: Human Molecular Genetics, vol. 27, no. 7, p. 1164–1173, 2018, ISSN: 1460-2083. @article{lenglet_dominant_2018,Acute intermittent porphyria (AIP) is a disease affecting the heme biosynthesis pathway caused by mutations of the hydroxymethylbilane synthase (HMBS) gene. AIP is thought to display autosomal dominant inheritance with incomplete penetrance. We evaluated the prevalence, penetrance and heritability of AIP, in families with the disease from the French reference center for porphyria (CFP) (602 overt patients; 1968 relatives) and the general population, using Exome Variant Server (EVS; 12 990 alleles) data. The pathogenicity of the 42 missense variants identified was assessed in silico, and in vitro, by measuring residual HMBS activity of the recombinant protein. The minimal estimated prevalence of AIP in the general population was 1/1299. Thus, 50 000 subjects would be expected to carry the AIP genetic trait in France. Penetrance was estimated at 22.9% in families with AIP, but at only 0.5-1% in the general population. Intrafamily correlation studies showed correlations to be strong overall and modulated by kinship and the area in which the person was living, demonstrating strong influences of genetic and environmental modifiers on inheritance. Null alleles were associated with a more severe phenotype and a higher penetrance than for other mutant alleles. In conclusion, the striking difference in the penetrance of HMBS mutations between the general population and the French AIP families suggests that AIP inheritance does not follow the classical autosomal dominant model, instead of being modulated by strong environmental and genetic factors independent from HMBS. An oligogenic inheritance model with environmental modifiers might better explain AIP penetrance and heritability. |
Pallet, Nicolas; Karras, Alexandre; Thervet, Eric; Gouya, Laurent; Karim, Zoubida; Puy, Hervé Porphyria and kidney diseases Article de journal Dans: Clinical Kidney Journal, vol. 11, no. 2, p. 191–197, 2018, ISSN: 2048-8505. @article{pallet_porphyria_2018,The kidneys, after the bone marrow and liver, are third in terms of the amounts of haem synthesized daily. Haem is incorporated into haemoproteins that are critical to renal physiology. In turn, disturbances in haem metabolism interfere with renal physiology and are tightly interrelated with kidney diseases. Acute intermittent porphyria causes kidney injury, whereas medical situations associated with end-stage renal disease, such as porphyrin accumulation, iron overload and hepatitis C, participate in the inhibition of uroporphyrinogen decarboxylase and predispose the individual to porphyria cutanea tarda. Even if some of these interactions have been known for a long time, the clinical situations associated with these interrelations have strikingly evolved over time with the advent of new therapeutic strategies for dialysis therapy and a better understanding of the pathophysiological mechanisms of porphyria-associated kidney disease. Physicians should be aware of these interactions. The aim of this review is to summarize the complex interactions between kidney physiology and pathology in the settings of porphyria and to emphasize their often-underestimated importance. |
Lefebvre, Thibaud; Reihani, Niloofar; Daher, Raed; de Villemeur, Thierry Billette; Belmatoug, Nadia; Rose, Christian; Colin-Aronovicz, Yves; Puy, Hervé; Le Van Kim, Caroline; Franco, Mélanie; Karim, Zoubida Involvement of hepcidin in iron metabolism dysregulation in Gaucher disease Article de journal Dans: Haematologica, vol. 103, no. 4, p. 587–596, 2018, ISSN: 1592-8721. @article{lefebvre_involvement_2018,Gaucher disease (GD) is an inherited deficiency of glucocerebrosidase leading to accumulation of glucosylceramide in tissues such as the spleen, liver, and bone marrow. The resulting lipid-laden macrophages lead to the appearance of "Gaucher cells". Anemia associated with an unexplained hyperferritinemia is a frequent finding in GD, but whether this pathogenesis is related to an iron metabolism disorder has remained unclear. To investigate this issue, we explored the iron status of a large cohort of 90 type I GD patients, including 66 patients treated with enzyme replacement therapy. Ten of the patients treated with enzyme replacement were followed up before and during treatment. Serum levels of hepcidin, the iron regulatory peptide, remained within the physiological range, while the transferrin saturation was slightly decreased in children. Inflammation-independent hyperferritinemia was found in 65% of the patients, and Perl's staining of the spleen and marrow smear revealed iron accumulation in Gaucher cells. Treated patients exhibited reduced hyperferritinemia, increased transferrin saturation and transiently increased systemic hepcidin. In addition, the hepcidin and ferritin correlation was markedly improved, and, in most patients, the hemoglobin level was normalized. To further explore eventual iron sequestration in macrophages, we produce a Gaucher cells model by treating the J774 macrophage cell line with a glucocerebrosidase inhibitor and showed induced local hepcidin and membrane retrieval of the iron exporter, ferroportin. These data reveal the involvement of Gaucher cells in abnormal iron sequestration, which may explain the mechanism of hyperferritinemia in GD patients. Local hepcidin-ferroportin interaction was involved in this pathogenesis. |
Hayder, Myriam; Garzoni, Matteo; Bochicchio, Davide; Caminade, Anne-Marie; Couderc, François; Ong-Meang, Varravaddheay; Davignon, Jean-Luc; Turrin, Cédric-Olivier; Pavan, Giovanni M; Poupot, Rémy Three-Dimensional Directionality Is a Pivotal Structural Feature for the Bioactivity of Azabisphosphonate-Capped Poly(PhosphorHydrazone) Nanodrug Dendrimers. Article de journal Dans: Biomacromolecules, vol. 19, no. 3, p. 712–720, 2018, ISSN: 1526-4602. @article{Hayder2018,Dendrimers are nanosized, nonlinear, hyperbranched polymers whose overall 3D shape is key for their biological activity. Poly(PhosphorHydrazone) (PPH) dendrimers capped with aza-bisphosphonate (ABP) end groups are known to have anti-inflammatory properties enabling the control of inflammatory diseases in different mouse models. Here we screen the anti-inflammatory activity of a series of PPH dendrimers bearing between 2 and 16 ABP end groups in a mouse model of arthritis and confront the biological results with atomistic simulations of the dendrimers. We show that only the PPH dendrimers capped with 10 and 12 ABP end groups can control the flare of the inflammatory disease. All-atom accelerated molecular dynamics simulations show that dendrimers with a low number of ABP end groups are directional but highly flexible/dynamic and have thereby limited efficiency in establishing multivalent interactions. The largest dendrimer appears as nondirectional, having 16 ABP end groups forming patches all over the dendrimer surface. Conversely, intermediate dendrimers having 10 or 12 ABP end groups reach the best compromise between the number of surface groups and their stable directional gathering, a real maximization of multivalency. |
Poupot, Rémy; Bergozza, Dylan; Fruchon, Séverine Nanoparticle-Based Strategies to Treat Neuro-Inflammation. Article de journal Dans: Materials (Basel, Switzerland), vol. 11, no. 2, p. 270, 2018, ISSN: 1996-1944. @article{Poupot2018a,Neuro-inflammation is a pivotal physio-pathological feature of brain disorders, including neurodegenerative diseases. As such, it is a relevant therapeutic target against which drugs have to be proposed. Targeting neuro-inflammation implies crossing the Blood-Brain Barrier (BBB) to reach the Central Nervous System (CNS). Engineered nanoparticles (ENPs) are promising candidates to carry and deliver drugs to the CNS by crossing the BBB. There are several strategies to design ENPs intended for crossing through the BBB. Herein, we first put nanotechnologies back in their historical context and introduce neuro-inflammation and its consequences in terms of public health. In a second part, we explain how ENPs can get access to the brain and review this area by highlighting recent papers in the field. Finally, after pointing out potential guidelines for preclinical studies involving ENPs, we conclude by opening the debate on the questions of nanosafety and toxicity of these ENPs and in particular on ecotoxicity related to regulatory issues and public concerns. |
Allas, Soraya; Caixàs, Assumpta; Poitou, Christine; Coupaye, Muriel; Thuilleaux, Denise; Lorenzini, Françoise; Diene, Gwenaëlle; Crinò, Antonino; Illouz, Frédéric; Grugni, Graziano; Potvin, Diane; Bocchini, Sarah; Delale, Thomas; Abribat, Thierry; Tauber, Maithé AZP-531, an unacylated ghrelin analog, improves food-related behavior in patients with Prader-Willi syndrome: A randomized placebo-controlled trial Article de journal Dans: PLoS ONE, vol. 13, no. 1, p. 1–19, 2018, ISSN: 19326203. @article{Allas2018,CONTEXT AND OBJECTIVE Prader-Willi syndrome (PWS) is characterized by early-onset hyperphagia and increased circulating levels of the orexigenic Acylated Ghrelin (AG) hormone with a relative deficit of Unacylated Ghrelin (UAG). AZP-531, a first-in-class UAG analog, was shown to inhibit the orexigenic effect of AG in animals, to improve glycemic control and decrease body weight in humans. We aimed to investigate the safety and efficacy of AZP-531 in patients with PWS for whom no approved treatment for hyperphagia is currently available. METHODS AND DESIGN Multi-center, randomized, double-blind, placebo-controlled trial. Forty-seven patients with genetically confirmed PWS and evidence of hyperphagia received daily subcutaneous injections of AZP-531 (3 and 4 mg for 50-70 kg and textgreater70 kg body weight, respectively) or matching placebo for 14 days. Assessments included adverse events, vital signs, safety laboratory tests, the Hyperphagia Questionnaire (HQ), patient-reported appetite, body composition and glycemic measures. RESULTS AZP-531 was well tolerated. There was a significant improvement with AZP-531 versus placebo in the mean total score, the 9-item score and the severity domain score of the HQ (p textless .05). The highest reduction in the total and 9-item scores was observed in AZP-531 subjects with the highest hyperphagia score at baseline. Findings were supported by a reduction in appetite scores observed with AZP-531 only. Body weight did not change in both groups while a significant reduction in waist circumference and fat mass was observed only with AZP-531. AZP-531 significantly decreased post-prandial glucose levels in a baseline glucose dependent fashion. CONCLUSIONS AZP-531 may constitute a new treatment strategy to improve hyperphagia and metabolic issues in patients with PWS. These findings support further investigation in longer-term clinical trials. |
Thuilleaux, Denise; Laurier, Virginie; Copet, Pierre; Tricot, Julie; Demeer, Geneviève; Mourre, Fabien; Tauber, Maithé; Jauregi, Joseba A model to characterize psychopathological features in adults with Prader-Willi syndrome Article de journal Dans: American Journal of Medical Genetics, Part A, vol. 176, no. 1, p. 41–47, 2018, ISSN: 15524833. @article{Thuilleaux2018,High prevalence of behavioral and psychiatric disorders in adults with Prader-Willi Syndrome (PWS) has been reported in last few years. However, data are confusing and often contradictory. In this article, we propose a model to achieve a better understanding of the psychopathological features in adults with PWS. The study is based on clinical observations of 150 adult inpatients, males and females. Non-parametric statistics were performed to analyse the association of psychopathological profiles with genotype, gender and age. We propose a model of psychiatric disorders in adults with PWS based on cognitive, emotional and behavioural issues. This model defines four psychopathological profiles: Basic, Impulsive, Compulsive, and Psychotic. The Basic profile is defined by traits and symptoms that are present in varying degrees in all persons with PWS. In our cohort, this Basic profile corresponds to 55% of the patients. The rest show, in addition to these characteristics, salient features of impulsivity (Impulsive profile, 19%), compulsivity (Compulsive profile, 7%), or psychosis (Psychotic profile, 19%). The analysis of factors associated with different profiles reveals an effect of genotype on Basic and Psychotic profiles (Deletion: 70% Basic, 9% Psychotic; Non-deletion: 23% Basic, 43% Psychotic) and a positive correlation between male sex and impulsivity, unmediated by sex hormone treatment. This is a clinical study, based on observation proposing an original model to understand the psychiatric and behavioural disorders in adults with PWS. Further studies are needed in order to test the validity of this model.Copyright textcopyright 2017 Wiley Periodicals, Inc. |
Puy, V.; Darwiche, W.; Trudel, S.; Gomila, C.; Lony, C.; Puy, L.; Lefebvre, T.; Vitry, S.; Boullier, A.; Karim, Z.; Ausseil, J. Predominant role of microglia in brain iron retention in Sanfilippo syndrome, a pediatric neurodegenerative disease Article de journal Dans: Glia, vol. 66, no. 8, p. 1709-1723, 2018, ISSN: 1098-1136 (Electronic) 0894-1491 (Linking). @article{RN1003, |
Rouillon, J.; Lefebvre, T.; Denard, J.; Puy, V.; Daher, R.; Ausseil, J.; Zocevic, A.; Fogel, P.; Peoc'h, K.; Wong, B.; Servais, L.; Voit, T.; Puy, H.; Karim, Z.; Svinartchouk, F. High urinary ferritin reflects myoglobin iron evacuation in DMD patients Article de journal Dans: Neuromuscul Disord, vol. 28, no. 7, p. 564-571, 2018, ISSN: 1873-2364 (Electronic) 0960-8966 (Linking). @article{RN1002, |
Tebani, A.; Abily-Donval, L.; Schmitz-Afonso, I.; Heron, B.; Piraud, M.; Ausseil, J.; Zerimech, F.; Gonzalez, B.; Marret, S.; Afonso, C.; Bekri, S. Unveiling metabolic remodeling in mucopolysaccharidosis type III through integrative metabolomics and pathway analysis Article de journal Dans: J Transl Med, vol. 16, no. 1, p. 248, 2018, ISSN: 1479-5876 (Electronic) 1479-5876 (Linking). @article{RN1001, |
Petillon, Camille; Hergesheimer, Rudolf; Puy, Hervé; Corcia, Philippe; Vourc'h, Patrick; Andres, Christian; Karim, Zoubida; Blasco, Hélène The Relevancy of Data Regarding the Metabolism of Iron to Our Understanding of Deregulated Mechanisms in ALS; Hypotheses and Pitfalls Article de journal Dans: Frontiers in Neuroscience, vol. 12, p. 1031, 2018, ISSN: 1662-4548. @article{petillon_relevancy_2018,Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease caused by the loss of motor neurons. Its etiology remains unknown, but several pathophysiological mechanisms are beginning to explain motor neuronal death, as well as oxidative stress. Iron accumulation has been observed in both sporadic and familial forms of ALS, including mouse models. Therefore, the dysregulation of iron metabolism could play a role in the pathological oxidative stress in ALS. Several studies have been undertaken to describe iron-related metabolic markers, in most cases focusing on metabolites in the bloodstream due to few available data in the central nervous system. Reports of accumulation of iron, high serum ferritin, and low serum transferrin levels in ALS patients have encouraged researchers to consider dysregulated iron metabolism as an integral part of ALS pathophysiology. However, it appears complicated to suggest a general mechanism due to the diversity of models and iron markers studied, including the lack of consensus among all of the studies. Regarding clinical study reports, most of them do not take into account confusion biases such as inflammation, renal dysfunction, and nutritional status. Furthermore, the iron regulatory pathways, particularly involving hepcidin, have not been thoroughly explored yet within the pathogenesis of iron overload in ALS. In this sense, it is also essential to explore the relation between iron overload and other ALS-related events, such as neuro-inflammation, protein aggregation, and iron-driven cell death, termed ferroptosis. In this review, we point out limits of the designs of certain studies that may prevent the understanding of the role of iron in ALS and discuss the relevance of the published data regarding the pathogenic impact of iron metabolism deregulation in this disease and the therapeutics targeting this pathway. |
Mabille, C.; Ruyssen-Witrand, A.; Degboe, Y.; Gennero, I.; Loiseau, H. A.; Roussel, M.; Hebraud, B.; Nigon, D.; Attal, M.; Laroche, M. DKK1 and sclerostin are early markers of relapse in multiple myeloma Article de journal Dans: Bone, vol. 113, p. 114-117, 2018, ISSN: 1873-2763 (Electronic) 1873-2763 (Linking), (Mabille, Charlotte Ruyssen-Witrand, Adeline Degboe, Yannick Gennero, Isabelle Loiseau, Herve Avet Roussel, Murielle Hebraud, Benjamin Nigon, Delphine Attal, Michel Laroche, Michel eng Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov't 2017/10/11 Bone. 2018 Aug;113:114-117. doi: 10.1016/j.bone.2017.10.004. Epub 2017 Oct 6.). @article{RN2137,Recent studies have shown that Dickkopf-related protein (DKK1) and sclerostin decrease when a complete response (CR) is obtained after chemotherapy in myeloma multiple (MM). To study variations in DKK1, sclerostin and P1NP in patients treated for MM, between complete response (CR) and relapse, we carried out a prospective study ancillary to the IFM 2009 protocol (IFM). The aim of IFM was to compare progression-free survival between patients treated with chemotherapy with or without transplantation. We selected 69 patients who reached CR and relapsed. We assayed by ELISA: DKK1, sclerostin and P1NP at 3 end points T1: CR, T2: 4 months before relapse and T3: relapse. There was a significant increase in DKK1 and sclerostin between T1, T2 and T3. (DKK1 medians (IQR): T1 = 30 pmol/l (20.4-41.1) |
Laroche, M.; Baradat, C.; Ruyssen-Witrand, A.; Degboe, Y. Variability of Denosumab densitometric response in postmenopausal osteoporosis Article de journal Dans: Rheumatol Int, vol. 38, no. 3, p. 461-466, 2018, ISSN: 1437-160X (Electronic) 0172-8172 (Linking), (Laroche, M Baradat, C Ruyssen-Witrand, A Degboe, Y eng Germany 2018/01/25 Rheumatol Int. 2018 Mar;38(3):461-466. doi: 10.1007/s00296-018-3929-0. Epub 2018 Jan 23.). @article{RN2138,The objective of our prospective study is to specify the variability of densitometric response to Denosumab, given in the second line, and to try to understand the reasons. All menopausal patients with primary osteoporosis, treated by Denosumab in our centre from 2014 to 2015, were included in this open prospective work. At T0, the patient's age, type of fracture, and previous treatments were collated. At T0 and T1, after 1 year of treatment by Dmab, a DXA of the spine and the hip and a determination of CTX were performed. Sixty-three patients aged 68.8 +/- 8.3 years were included. The median number of treatments prescribed for osteoporosis before switch to Denosumab was 2.4. The median duration of these treatments was 7.2 years. At T1, CTX was less than 33 pg/ml (minimum threshold for our assay kit) in all patients. The median BMD in the spine increased by + 5.44% compared to T0. 14 patients in the upper quartile had a median BMD gain in the spine of + 11.07%. Fourteen patients in the lower quartile had a median BMD gain in the spine of + 0.6%. Only the duration of previous treatments, which was greater in the non-responder group, differed between these two groups. In the total cohort, the spinal densitometric gain was negatively correlated with the age of the patient at baseline (p = 0.04), the duration of previous treatment (p = 0.02), and positively with the CTX level (p = 0.05). The Dmab densitometric response is highly variable, partly explained by the duration of previous treatments and the level of bone resorption at initiation of treatment. |
2017 |
Daher, Raed; Manceau, Hana; Karim, Zoubida Iron metabolism and the role of the iron-regulating hormone hepcidin in health and disease Article de journal Dans: Presse Medicale (Paris, France: 1983), vol. 46, no. 12 Pt 2, p. e272–e278, 2017, ISSN: 2213-0276. @article{daher_iron_2017,Although iron is vital, its free form is likely to be involved in oxidation-reduction reactions, leading to the formation of free radicals and oxidative stress. Living organisms have developed protein systems to transport free iron through the cell membranes and biological fluids and store it in a non-toxic and readily mobilizable form to avoid iron toxicity. Hepcidin plays a crucial role in maintaining iron homeostasis. Hepcidin expression is directly regulated by variations in iron intake and its repression leads to an increase in bioavailable serum iron level. However, in pathological situations, prolonged repression often leads to pathological iron overload. In this review, we describe the different molecular mechanisms responsible for the maintenance of iron metabolism and the consequences of iron overload. Indeed, genetic hemochromatosis and post-transfusional siderosis are the two main conditions responsible for iron overload. Long-term iron overload is deleterious, and treatment relies on venesection therapy for genetic hemochromatosis and chelation therapy for iron overload resulting from multiple transfusions. |
Fruchon, Séverine; Poupot, Rémy Pro-Inflammatory Versus Anti-Inflammatory Effects of Dendrimers: The Two Faces of Immuno-Modulatory Nanoparticles. Article de journal Dans: Nanomaterials (Basel, Switzerland), vol. 7, no. 9, p. 251, 2017, ISSN: 2079-4991. @article{Fruchon2017,Dendrimers are soft matter, hyperbranched, and multivalent nanoparticles whose synthesis theoretically affords monodisperse compounds. They are built from a core on which one or several successive series of branches are engrafted in an arborescent way. At the end of the synthesis, the tunable addition of surface groups gives birth to multivalent nano-objects which are generally intended for a specific use. For these reasons, dendrimers have received a lot of attention from biomedical researchers. In particular, some of us have demonstrated that dendrimers can be intrinsically drug-candidate for the treatment of inflammatory disorders, amongst others, using relevant preclinical animal models. These anti-inflammatory dendrimers are innovative in the pharmaceutical field. More recently, it has appeared that some dendrimers (even among those which have been described as anti-inflammatory) can promote inflammatory responses in non-diseased animals. The main corpus of this concise review is focused on the reports which describe anti-inflammatory properties of dendrimers in vivo, following which we review the few recent articles that show pro-inflammatory effects of our favorite molecules, to finally discuss this duality in immuno-modulation which has to be taken into account for the preclinical and clinical developments of dendrimers. |
Bakouh, Naziha; Bellanca, Sebastiano; Nyboer, Britta; Moliner Cubel, Sonia; Karim, Zoubida; Sanchez, Cecilia P.; Stein, Wilfred D.; Planelles, Gabrielle; Lanzer, Michael Iron is a substrate of the Plasmodium falciparum chloroquine resistance transporter PfCRT in Xenopus oocytes Article de journal Dans: The Journal of Biological Chemistry, vol. 292, no. 39, p. 16109–16121, 2017, ISSN: 1083-351X. @article{bakouh_iron_2017,The chloroquine resistance transporter of the human malaria parasite Plasmodium falciparum, PfCRT, is an important determinant of resistance to several quinoline and quinoline-like antimalarial drugs. PfCRT also plays an essential role in the physiology of the parasite during development inside erythrocytes. However, the function of this transporter besides its role in drug resistance is still unclear. Using electrophysiological and flux experiments conducted on PfCRT-expressing Xenopus laevis oocytes, we show here that both wild-type PfCRT and a PfCRT variant associated with chloroquine resistance transport both ferrous and ferric iron, albeit with different kinetics. In particular, we found that the ability to transport ferrous iron is reduced by the specific polymorphisms acquired by the PfCRT variant as a result of chloroquine selection. We further show that iron and chloroquine transport via PfCRT is electrogenic. If these findings in the Xenopus model extend to P. falciparum in vivo, our data suggest that PfCRT might play a role in iron homeostasis, which is essential for the parasite's development in erythrocytes. |
Van Aelst, Lucas N. L.; Abraham, Marjorie; Sadoune, Malha; Lefebvre, Thibaud; Manivet, Philippe; Logeart, Damien; Launay, Jean-Marie; Karim, Zoubida; Puy, Hervé; Cohen-Solal, Alain Iron status and inflammatory biomarkers in patients with acutely decompensated heart failure: early in-hospital phase and 30-day follow-up Article de journal Dans: European Journal of Heart Failure, vol. 19, no. 8, p. 1075–1076, 2017, ISSN: 1879-0844. @article{van_aelst_iron_2017, |
Millot, Sarah; Delaby, Constance; Moulouel, Boualem; Lefebvre, Thibaud; Pilard, Nathalie; Ducrot, Nicolas; Ged, Cécile; Lettéron, Philippe; de Franceschi, Lucia; Deybach, Jean Charles; Beaumont, Carole; Gouya, Laurent; De Verneuil, Hubert; Lyoumi, Saïd; Puy, Hervé; Karim, Zoubida Hemolytic anemia repressed hepcidin level without hepatocyte iron overload: lesson from Günther disease model Article de journal Dans: Haematologica, vol. 102, no. 2, p. 260–270, 2017, ISSN: 1592-8721. @article{millot_hemolytic_2017,Hemolysis occurring in hematologic diseases is often associated with an iron loading anemia. This iron overload is the result of a massive outflow of hemoglobin into the bloodstream, but the mechanism of hemoglobin handling has not been fully elucidated. Here, in a congenital erythropoietic porphyria mouse model, we evaluate the impact of hemolysis and regenerative anemia on hepcidin synthesis and iron metabolism. Hemolysis was confirmed by a complete drop in haptoglobin, hemopexin and increased plasma lactate dehydrogenase, an increased red blood cell distribution width and osmotic fragility, a reduced half-life of red blood cells, and increased expression of heme oxygenase 1. The erythropoiesis-induced Fam132b was increased, hepcidin mRNA repressed, and transepithelial iron transport in isolated duodenal loops increased. Iron was mostly accumulated in liver and spleen macrophages but transferrin saturation remained within the normal range. The expression levels of hemoglobin-haptoglobin receptor CD163 and hemopexin receptor CD91 were drastically reduced in both liver and spleen, resulting in heme- and hemoglobin-derived iron elimination in urine. In the kidney, the megalin/cubilin endocytic complex, heme oxygenase 1 and the iron exporter ferroportin were induced, which is reminiscent of significant renal handling of hemoglobin-derived iron. Our results highlight ironbound hemoglobin urinary clearance mechanism and strongly suggest that, in addition to the sequestration of iron in macrophages, kidney may play a major role in protecting hepatocytes from iron overload in chronic hemolysis. |
Burnett, L. C.; LeDuc, C. A.; Sulsona, C. R.; Paull, D.; Rausch, R.; Eddiry, S.; Carli, J. F.; Morabito, M. V.; Skowronski, A. A.; Hubner, G.; Zimmer, M.; Wang, L.; Day, R.; Levy, B.; Fennoy, I.; Dubern, B.; Poitou, C.; Clement, K.; Butler, M. G.; Rosenbaum, M.; Salles, J. P.; Tauber, M.; Driscoll, D. J.; Egli, D.; Leibel, R. L. Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome Article de journal Dans: J Clin Invest, vol. 127, no. 1, p. 293-305, 2017, ISSN: 1558-8238 (Electronic) 0021-9738 (Linking). @article{RN2b, |
Tauber, Maïthé; Boulanouar, Kader; Diene, Gwenaelle; Çabal-Berthoumieu, Sophie; Ehlinger, Virginie; Fichaux-Bourin, Pascale; Molinas, Catherine; Faye, Sandy; Valette, Marion; Pourrinet, Jeanne; Cessans, Catie; Viaux-Sauvelon, Sylvie; Bascoul, Céline; Guedeney, Antoine; Delhanty, Patric; Geenen, Vincent; Martens, Henri; Muscatelli, Françoise; Cohen, David; Consoli, Angèle; Payoux, Pierre; Arnaud, Catherine; Salles, Jean-Pierre The Use of Oxytocin to Improve Feeding and Social Skills in Infants With Prader–Willi Syndrome Article de journal Dans: PEDIATRICS, vol. 139, no. 2, p. e2 0162976, 2017. @article{RN10b, |
Bodet, P. E.; Salard, I.; Przybylski, C.; Gonnet, F.; Gomila, C.; Ausseil, J.; Daniel, R. Efficient recovery of glycosaminoglycan oligosaccharides from polyacrylamide gel electrophoresis combined with mass spectrometry analysis Article de journal Dans: Anal Bioanal Chem, vol. 409, no. 5, p. 1257-1269, 2017, ISSN: 1618-2650 (Electronic) 1618-2642 (Linking). @article{RN1011, |
Daher, I.; Le Dieu-Lugon, B.; Dourmap, N.; Lecuyer, M.; Ramet, L.; Gomila, C.; Ausseil, J.; Marret, S.; Leroux, P.; Roy, V.; El Mestikawy, S.; Daumas, S.; Gonzalez, B.; Leroux-Nicollet, I.; Cleren, C. Magnesium Sulfate Prevents Neurochemical and Long-Term Behavioral Consequences of Neonatal Excitotoxic Lesions: Comparison Between Male and Female Mice Article de journal Dans: J Neuropathol Exp Neurol, vol. 76, no. 10, p. 883-897, 2017, ISSN: 1554-6578 (Electronic) 0022-3069 (Linking). @article{RN1007, |
Hodroge, A.; Trecherel, E.; Cornu, M.; Darwiche, W.; Mansour, A.; Ait-Mohand, K.; Verissimo, T.; Gomila, C.; Schembri, C.; Da Nascimento, S.; Elboutachfaiti, R.; Boullier, A.; Lorne, E.; Courtois, J.; Petit, E.; Toumieux, S.; Kovensky, J.; Sonnet, P.; Massy, Z. A.; Kamel, S.; Rossi, C.; Ausseil, J. Oligogalacturonic Acid Inhibits Vascular Calcification by Two Mechanisms: Inhibition of Vascular Smooth Muscle Cell Osteogenic Conversion and Interaction With Collagen Article de journal Dans: Arterioscler Thromb Vasc Biol, vol. 37, no. 7, p. 1391-1401, 2017, ISSN: 1524-4636 (Electronic) 1079-5642 (Linking). @article{RN1009, |
Tardieu, M.; Zerah, M.; Gougeon, M. L.; Ausseil, J.; de Bournonville, S.; Husson, B.; Zafeiriou, D.; Parenti, G.; Bourget, P.; Poirier, B.; Furlan, V.; Artaud, C.; Baugnon, T.; Roujeau, T.; Crystal, R. G.; Meyer, C.; Deiva, K.; Heard, J. M. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: an uncontrolled phase 1/2 clinical trial Article de journal Dans: Lancet Neurol, vol. 16, no. 9, p. 712-720, 2017, ISSN: 1474-4465 (Electronic) 1474-4422 (Linking). @article{RN1008, |
Tebani, A.; Schmitz-Afonso, I.; Abily-Donval, L.; Heron, B.; Piraud, M.; Ausseil, J.; Brassier, A.; De Lonlay, P.; Zerimech, F.; Vaz, F. M.; Gonzalez, B. J.; Marret, S.; Afonso, C.; Bekri, S. Urinary metabolic phenotyping of mucopolysaccharidosis type I combining untargeted and targeted strategies with data modeling Article de journal Dans: Clin Chim Acta, vol. 475, p. 7-14, 2017, ISSN: 1873-3492 (Electronic) 0009-8981 (Linking). @article{RN1006, |
Yien, Yvette Y.; Ducamp, Sarah; van der Vorm, Lisa N.; Kardon, Julia R.; Manceau, Hana; Kannengiesser, Caroline; Bergonia, Hector A.; Kafina, Martin D.; Karim, Zoubida; Gouya, Laurent; Baker, Tania A.; Puy, Hervé; Phillips, John D.; Nicolas, Gaël; Paw, Barry H. Mutation in human CLPX elevates levels of δ-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria Article de journal Dans: Proceedings of the National Academy of Sciences of the United States of America, vol. 114, no. 38, p. E8045–E8052, 2017, ISSN: 1091-6490. @article{yien_mutation_2017,Loss-of-function mutations in genes for heme biosynthetic enzymes can give rise to congenital porphyrias, eight forms of which have been described. The genetic penetrance of the porphyrias is clinically variable, underscoring the role of additional causative, contributing, and modifier genes. We previously discovered that the mitochondrial AAA+ unfoldase ClpX promotes heme biosynthesis by activation of δ-aminolevulinate synthase (ALAS), which catalyzes the first step of heme synthesis. CLPX has also been reported to mediate heme-induced turnover of ALAS. Here we report a dominant mutation in the ATPase active site of human CLPX, p.Gly298Asp, that results in pathological accumulation of the heme biosynthesis intermediate protoporphyrin IX (PPIX). Amassing of PPIX in erythroid cells promotes erythropoietic protoporphyria (EPP) in the affected family. The mutation in CLPX inactivates its ATPase activity, resulting in coassembly of mutant and WT protomers to form an enzyme with reduced activity. The presence of low-activity CLPX increases the posttranslational stability of ALAS, causing increased ALAS protein and ALA levels, leading to abnormal accumulation of PPIX. Our results thus identify an additional molecular mechanism underlying the development of EPP and further our understanding of the multiple mechanisms by which CLPX controls heme metabolism. |
de Montalembert, Mariane; Ribeil, Jean-Antoine; Brousse, Valentine; Guerci-Bresler, Agnes; Stamatoullas, Aspasia; Vannier, Jean-Pierre; Dumesnil, Cécile; Lahary, Agnès; Touati, Mohamed; Bouabdallah, Krimo; Cavazzana, Marina; Chauzit, Emmanuelle; Baptiste, Amandine; Lefebvre, Thibaud; Puy, Hervé; Elie, Caroline; Karim, Zoubida; Ernst, Olivier; Rose, Christian Cardiac iron overload in chronically transfused patients with thalassemia, sickle cell anemia, or myelodysplastic syndrome Article de journal Dans: PloS One, vol. 12, no. 3, p. e0172147, 2017, ISSN: 1932-6203. @article{de_montalembert_cardiac_2017,The risk and clinical significance of cardiac iron overload due to chronic transfusion varies with the underlying disease. Cardiac iron overload shortens the life expectancy of patients with thalassemia, whereas its effect is unclear in those with myelodysplastic syndromes (MDS). In patients with sickle cell anemia (SCA), iron does not seem to deposit quickly in the heart. Our primary objective was to assess through a multicentric study the prevalence of cardiac iron overload, defined as a cardiovascular magnetic resonance T2*textless20 ms, in patients with thalassemia, SCA, or MDS. Patient inclusion criteria were an accurate record of erythrocyte concentrates (ECs) received, a transfusion history textgreater8 ECs in the past year, and age older than 6 years. We included from 9 centers 20 patients with thalassemia, 41 with SCA, and 25 with MDS in 2012-2014. Erythrocytapharesis did not consistently prevent iron overload in patients with SCA. Cardiac iron overload was found in 3 (15%) patients with thalassemia, none with SCA, and 4 (16%) with MDS. The liver iron content (LIC) ranged from 10.4 to 15.2 mg/g dry weight, with no significant differences across groups (P = 0.29). Abnormal T2* was not significantly associated with any of the measures of transfusion or chelation. Ferritin levels showed a strong association with LIC. Non-transferrin-bound iron was high in the thalassemia and MDS groups but low in the SCA group (Ptextless0.001). Hepcidin was low in thalassemia, normal in SCA, and markedly elevated in MDS (Ptextless0.001). Two mechanisms may explain that iron deposition largely spares the heart in SCA: the high level of erythropoiesis recycles the iron and the chronic inflammation retains iron within the macrophages. Thalassemia, in contrast, is characterized by inefficient erythropoiesis, unable to handle free iron. Iron accumulation varies widely in MDS syndromes due to the competing influences of abnormal erythropoiesis, excess iron supply, and inflammation. |
Tchernitchko, Dimitri; Tavernier, Quentin; Lamoril, Jérôme; Schmitt, Caroline; Talbi, Neila; Lyoumi, Said; Robreau, Anne-Marie; Karim, Zoubida; Gouya, Laurent; Thervet, Eric; Karras, Alexandre; Puy, Hervé; Pallet, Nicolas A Variant of Peptide Transporter 2 Predicts the Severity of Porphyria-Associated Kidney Disease Article de journal Dans: Journal of the American Society of Nephrology: JASN, vol. 28, no. 6, p. 1924–1932, 2017, ISSN: 1533-3450. @article{tchernitchko_variant_2017,CKD occurs in most patients with acute intermittent porphyria (AIP). During AIP, δ-aminolevulinic acid (ALA) accumulates and promotes tubular cell death and tubulointerstitial damage. The human peptide transporter 2 (PEPT2) expressed by proximal tubular cells mediates the reabsorption of ALA, and variants of PEPT2 have different affinities for ALA. We tested the hypothesis that PEPT2 genotypes affect the severity and prognosis of porphyria-associated kidney disease. We analyzed data from 122 individuals with AIP who were followed from 2003 to 2013 and genotyped for PEPT2 At last follow-up, carriers of the PEPT2*1*1 genotype (higher affinity variant) exhibited worse renal function than carriers of the lower affinity variants PEPT2*1/*2 and PEPT2*2/*2 (mean±SD eGFR: 54.4±19.1, 66.6±23.8, and 78.1±19.9 ml/min per 1.73 m2, respectively). Change in eGFR (mean±SD) over the 10-year period was -11.0±3.3, -2.4±1.9, and 3.4±2.6 ml/min per 1.73 m2 for PEPT2*1/*1, PEPT2*1*2, and PEPT*2*2*2 carriers, respectively. At the end of follow-up, 68% of PEPT2*1*1 carriers had an eGFRtextless60 ml/min per 1.73 m2, compared with 37% of PEPT2*1*2 carriers and 15% of PEPT2*2*2 carriers. Multiple regression models including all confounders indicated that the PEPT2*1*1 genotype independently associated with an eGFRtextless60 ml/min per 1.73 m2 (odds ratio, 6.85; 95% confidence interval, 1.34 to 46.20) and an annual decrease in eGFR of textgreater1 ml/min per 1.73 m2 (odds ratio, 3.64; 95% confidence interval, 1.37 to 9.91). Thus, a gene variant is predictive of the severity of a chronic complication of AIP. The therapeutic value of PEPT2 inhibitors in preventing porphyria-associated kidney disease warrants investigation. |
Degboe, Y.; Eischen, M.; Nigon, D.; Apoil, P. A.; Mailhol, C.; Tournier, E.; Laurent, C.; Hanssens, K.; Hermine, O.; Paul, C.; Laroche, M.; Bulai-Livideanu, C. Prevalence and risk factors for fragility fracture in systemic mastocytosis Article de journal Dans: Bone, vol. 105, p. 219-225, 2017, ISSN: 1873-2763 (Electronic) 1873-2763 (Linking), (Degboe, Yannick Eischen, Marine Nigon, Delphine Apoil, Pol-Andre Mailhol, Claire Tournier, Emilie Laurent, Camille Hanssens, Katia Hermine, Olivier Paul, Carle Laroche, Michel Bulai-Livideanu, Cristina eng 2017/09/19 Bone. 2017 Dec;105:219-225. doi: 10.1016/j.bone.2017.09.005. Epub 2017 Sep 14.). @article{RN2140,OBJECTIVES: Systemic mastocytosis (SM) is characterized by the accumulation of mast cells in tissues other than the skin. Bone involvement although frequent has not been thoroughly evaluated. Primary objective was to determine risk factors associated with fragility fractures (FF) in SM. Secondary objectives were to evaluate the ability of bone marrow tryptase (BMT) level to identify patients with FF, and to describe bone involvement in SM. METHODS: We analyzed retrospectively all consecutive patients seen in our expert center, with a diagnosis of SM according to the 2001 WHO criteria, and with complete bone assessment. We collected data about lifetime fractures, types of cutaneous manifestations, degranulation symptoms, blood and BMT levels, bone mineral density assessed by densitometry and KIT mutation. We performed a univariate analysis investigating the factors associated with FF and then a logistic multivariable regression analysis. We assessed the ability of bone marrow tryptase to identify patients with FF. RESULTS: Eighty-nine patients with SM were included. Thirty-six patients (40.4%) suffered from osteoporosis and twenty-five (28.1%) experienced lifetime FF. Univariate analysis identified age at diagnosis and disease onset, presence of telangiectasia macularis eruptiva perstans, digestive symptoms, mast cells activation symptoms, elevated BMT, low femoral and lumbar BMD, as associated with FF. Multivariate analysis identified elevated BMT, low femoral T score and older age at diagnosis as independently associated with FF. CONCLUSIONS: Low femoral T-score, BMT level, and older age at diagnosis are markers associated with FF in SM. BMT may represent an important biomarker to predict FF in SM patients. |
2016 |
Karim, Zoubida; Puy, Hervé; Beaumont, Carole; Gouya, Laurent; Kannengiesser, Caroline Reply Article de journal Dans: Gastroenterology, vol. 151, no. 4, p. 771–772, 2016, ISSN: 1528-0012. @article{karim_reply_2016, |
Daher, Raed; Kannengiesser, Caroline; Houamel, Dounia; Lefebvre, Thibaud; Bardou-Jacquet, Edouard; Ducrot, Nicolas; de Kerguenec, Caroline; Jouanolle, Anne-Marie; Robreau, Anne-Marie; Oudin, Claire; Le Gac, Gerald; Moulouel, Boualem; Loustaud-Ratti, Veronique; Bedossa, Pierre; Valla, Dominique; Gouya, Laurent; Beaumont, Carole; Brissot, Pierre; Puy, Hervé; Karim, Zoubida; Tchernitchko, Dimitri Heterozygous Mutations in BMP6 Pro-peptide Lead to Inappropriate Hepcidin Synthesis and Moderate Iron Overload in Humans Article de journal Dans: Gastroenterology, vol. 150, no. 3, p. 672–683.e4, 2016, ISSN: 1528-0012. @article{daher_heterozygous_2016,BACKGROUND & AIMS: Hereditary hemochromatosis is a heterogeneous group of genetic disorders characterized by parenchymal iron overload. It is caused by defective expression of liver hepcidin, the main regulator of iron homeostasis. Iron stimulates the gene encoding hepcidin (HAMP) via the bone morphogenetic protein (BMP)6 signaling to SMAD. Although several genetic factors have been found to cause late-onset hemochromatosis, many patients have unexplained signs of iron overload. We investigated BMP6 function in these individuals. METHODS: We sequenced the BMP6 gene in 70 consecutive patients with a moderate increase in serum ferritin and liver iron levels who did not carry genetic variants associated with hemochromatosis. We searched for BMP6 mutations in relatives of 5 probands and in 200 healthy individuals (controls), as well as in 2 other independent cohorts of hyperferritinemia patients. We measured serum levels of hepcidin by liquid chromatography-tandem mass spectrometry and analyzed BMP6 in liver biopsy specimens from patients by immunohistochemistry. The functions of mutant and normal BMP6 were assessed in transfected cells using immunofluorescence, real-time quantitative polymerase chain reaction, and immunoblot analyses. RESULTS: We identified 3 heterozygous missense mutations in BMP6 (p.Pro95Ser, p.Leu96Pro, and p.Gln113Glu) in 6 unrelated patients with unexplained iron overload (9% of our cohort). These mutations were detected in less than 1% of controls. p.Leu96Pro also was found in 2 patients from the additional cohorts. Family studies indicated dominant transmission. Serum levels of hepcidin were inappropriately low in patients. A low level of BMP6, compared with controls, was found in a biopsy specimen from 1 patient. In cell lines, the mutated residues in the BMP6 propeptide resulted in defective secretion of BMP6; reduced signaling via SMAD1, SMAD5, and SMAD8; and loss of hepcidin production. CONCLUSIONS: We identified 3 heterozygous missense mutations in BMP6 in patients with unexplained iron overload. These mutations lead to loss of signaling to SMAD proteins and reduced hepcidin production. These mutations might increase susceptibility to mild-to-moderate late-onset iron overload. |
Houamel, Dounia; Ducrot, Nicolas; Lefebvre, Thibaud; Daher, Raed; Moulouel, Boualem; Sari, Marie-Agnes; Letteron, Philippe; Lyoumi, Said; Millot, Sarah; Tourret, Jerome; Bouvet, Odile; Vaulont, Sophie; Vandewalle, Alain; Denamur, Erick; Puy, Hervé; Beaumont, Carole; Gouya, Laurent; Karim, Zoubida Hepcidin as a Major Component of Renal Antibacterial Defenses against Uropathogenic Escherichia coli Article de journal Dans: Journal of the American Society of Nephrology: JASN, vol. 27, no. 3, p. 835–846, 2016, ISSN: 1533-3450. @article{houamel_hepcidin_2016,The iron-regulatory peptide hepcidin exhibits antimicrobial activity. Having previously shown hepcidin expression in the kidney, we addressed its role in urinary tract infection (UTI), which remains largely unknown. Experimental UTI was induced in wild-type (WT) and hepcidin-knockout (Hepc-/-) mice using the uropathogenic Escherichia coli CFT073 strain. Compared with infected WT mice, infected Hepc-/- mice showed a dramatic increase in renal bacterial load. Moreover, bacterial invasion was significantly dampened by the pretreatment of WT mice with hepcidin. Infected Hepc-/- mice exhibited decreased iron accumulation in the renal medulla and significant attenuation of the renal inflammatory response. Notably, we demonstrated in vitro bacteriostatic activity of hepcidin against CFT073. Furthermore, CFT073 repressed renal hepcidin, both in vivo and in cultured renal cells, and reduced phosphorylation of SMAD kinase in vivo, suggesting a bacterial strategy to escape the antimicrobial activities of hepcidin. In conclusion, we provide new mechanisms by which hepcidin contributes to renal host defense and suggest that targeting hepcidin offers a strategy to prevent bacterial invasion. |
Puy, Hervé; Manceau, Hana; Karim, Zoubida; Kan-Nengiesser, Caroline Rare microcytic anemias Article de journal Dans: Bulletin De l'Academie Nationale De Medecine, vol. 200, no. 2, p. 335–347, 2016, ISSN: 0001-4079. @article{puy_rare_2016,Microcytic anemia is often due to disorder of globin genes. Here, we focus on rare monogenic microcytic anemias, We describe the diferent congenital forms that are due to mutations in genes implicated in iron homeostasis, the heme biosynthesis pathway, the cluster Fe-S biosynthesis pathway andmitochondrial proteins biosynthesis pathway. Among rare congenital microcytic anemias, most frequent forms are non syndromic sideroblastic anemias and iron refractory iron deficiency anemias (IRIDA). Sideroblastic anemias is characterized by mitochondrial iron overload and presence of ring sideroblasts in patient bone marrow.. IRIDA results from bi-allelic mutations of TMPRSS6 gene encoding Matriptase-2. Matriptase-2 protein is a transmembrane serine protease that plays an essential role in down-regulating hepcidin, the key regulator of iron homeostasis. The next generation sequencing would be helpful to identify the genes implicated in unexplained rare anemias. |
Burnett, L. C.; LeDuc, C. A.; Sulsona, C. R.; Paull, D.; Eddiry, S.; Levy, B.; Salles, J. P.; Tauber, M.; Driscoll, D. J.; Egli, D.; Leibel, R. L. Induced pluripotent stem cells (iPSC) created from skin fibroblasts of patients with Prader-Willi syndrome (PWS) retain the molecular signature of PWS Article de journal Dans: Stem Cell Res, vol. 17, no. 3, p. 526-530, 2016, ISSN: 1876-7753 (Electronic) 1873-5061 (Linking). @article{RN1b, |
Boyer, J. F.; Baron, M.; Constantin, A.; Degboe, Y.; Cantagrel, A.; Davignon, J. L. Anti-TNF certolizumab pegol induces antioxidant response in human monocytes via reverse signaling Article de journal Dans: Arthritis Res Ther, vol. 18, p. 56, 2016, ISSN: 1478-6362 (Electronic) 1478-6354 (Linking). @article{RN11b, |
Ielasi, F.; Ledall, J.; Anes, A. P.; Fruchon, S.; Caminade, A. M.; Poupot, R.; Turrin, C. O.; Blanzat, M. Influence of PPH dendrimers' surface functions on the activation of human monocytes: a study of their interactions with pure lipid model systems Article de journal Dans: Phys Chem Chem Phys, vol. 18, no. 31, p. 21871-80, 2016, ISSN: 1463-9084 (Electronic) 1463-9076 (Linking). @article{RN24b, |
Poupot, M.; Turrin, C. O.; Caminade, A. M.; Fournie, J. J.; Attal, M.; Poupot, R.; Fruchon, S. Poly(phosphorhydrazone) dendrimers: yin and yang of monocyte activation for human NK cell amplification applied to immunotherapy against multiple myeloma Article de journal Dans: Nanomedicine, vol. 12, no. 8, p. 2321-2330, 2016, ISSN: 1549-9642 (Electronic) 1549-9634 (Linking). @article{RN36, |
Puy, V.; Zmudka-Attier, J.; Capel, C.; Bouzerar, R.; Serot, J. M.; Bourgeois, A. M.; Ausseil, J.; Baledent, O. Interactions between Flow Oscillations and Biochemical Parameters in the Cerebrospinal Fluid Article de journal Dans: Front Aging Neurosci, vol. 8, p. 154, 2016, ISSN: 1663-4365 (Print) 1663-4365 (Linking). @article{RN1013, |
Deschemin, Jean-Christophe; Noordine, Marie-Louise; Remot, Aude; Willemetz, Alexandra; Afif, Clément; Canonne-Hergaux, François; Langella, Philippe; Karim, Zoubida; Vaulont, Sophie; Thomas, Muriel; Nicolas, Gaël The microbiota shifts the iron sensing of intestinal cells Article de journal Dans: FASEB journal: official publication of the Federation of American Societies for Experimental Biology, vol. 30, no. 1, p. 252–261, 2016, ISSN: 1530-6860. @article{deschemin_microbiota_2016,The amount of iron in the diet directly influences the composition of the microbiota. Inversely, the effects of the microbiota on iron homeostasis have been little studied. So, we investigate whether the microbiota itself may alter host iron sensing. Duodenal cytochrome b and divalent metal transporter 1, involved in apical iron uptake, are 8- and 10-fold, respectively, more abundant in the duodenum of germ-free (GF) mice than in mice colonized with a microbiota. In contrast, the luminal exporter ferroportin is 2-fold less abundant in GF. The overall signature of microbiota on iron-related proteins is similar in the colon. The colonization does not modify systemic parameters as plasma transferrin saturation (20%), plasma ferritin (150 ng/L), and liver (85 µg/g) iron load. Commensal organisms (Bacteroides thetaiotaomicron VPI-5482 and Faecalibacterium prausnitzii A2-165) and a probiotic strain (Streptococcus thermophilus LMD-9) led to up to 12-fold induction of ferritin in colon. Our data suggest that the intestinal cells of GF mice are depleted of iron and that following colonization, the epithelial cells favor iron storage. This study is the first to demonstrate that gut microbes induce a specific iron-related protein signature, highlighting new aspects of the crosstalk between the microbiota and the intestinal epithelium. |
2015 |
Lefebvre, Thibaud; Dessendier, Nathalie; Houamel, Dounia; Ialy-Radio, Nathalie; Kannengiesser, Caroline; Manceau, Hana; Beaumont, Carole; Nicolas, Gael; Gouya, Laurent; Puy, Hervé; Karim, Zoubida LC-MS/MS method for hepcidin-25 measurement in human and mouse serum: clinical and research implications in iron disorders Article de journal Dans: Clinical Chemistry and Laboratory Medicine, vol. 53, no. 10, p. 1557–1567, 2015, ISSN: 1437-4331. @article{lefebvre_lc-msms_2015,BACKGROUND: The peptide hepcidin plays a central role in regulating dietary iron absorption and body iron distribution. This 25-amino acid hormone is produced and secreted predominantly by hepatocytes. Hepcidin has been suggested as a promising diagnostic marker for iron-related disorders. However, its accurate quantification for clinical use remains so far challenging. In this report we describe a highly specific and quantitative serum hepcidin method using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). MATERIAL: The analytical validation included the determination of the limit of detection, of quantification, repeatability, reproducibility and linearity. This assay was developed for human and mouse hepcidin. The human assay was performed on serum patients with unexplained microcytic anemia. We applied our LC-MS/MS method for quantifying hepcidin-1 in mouse in various conditions: inflammation, hemolytic anemia, Hamp-1, Hjv and Hfe KO mice. RESULTS: We show that the LC-MS/MS is suitable for accurate determination of hepcidin-25 in clinical samples, thereby representing a useful tool for the clinical diagnosis and follow-up of iron-related diseases. In mouse, a strong correlation between hepatic Hamp-1 mRNA expression and serum hepcidin-1 levels was found (r=0.88; p=0.0002) and the expected variations in mouse models of iron disorders were observed. CONCLUSIONS: Therefore, we propose this adaptive LC-MS/MS method as a suitable method for accurate determination of hepcidin-25 in clinical samples and as a major tool contributing to the clinical diagnosis, follow-up and management of iron-related disorders. It also opens new avenues to measure hepcidin in animal models without interspecies antigenic limitations. |
Karim, Zoubida; Lyoumi, Said; Nicolas, Gael; Deybach, Jean-Charles; Gouya, Laurent; Puy, Hervé Porphyrias: A 2015 update Article de journal Dans: Clinics and Research in Hepatology and Gastroenterology, vol. 39, no. 4, p. 412–425, 2015, ISSN: 2210-741X. @article{karim_porphyrias_2015,The hereditary porphyrias comprise a group of eight metabolic disorders of the heme biosynthesis pathway. Each porphyria is caused by abnormal function at a separate enzymatic step resulting in a specific accumulation of heme precursors. Porphyrias are classified as hepatic or erythropoietic, based on the organ system in which heme precursors (δ-aminolevulinic acid [ALA], porphobilinogen and porphyrins) are overproduced. Clinically, porphyrias are characterized by acute neurovisceral symptoms, skin lesions or both. However, most if not all the porphyrias impair hepatic or gastrointestinal function. Acute hepatic porphyrias present with severe abdominal pain, nausea, constipation, confusion and seizure, which may be life threatening, and patients are at risk of hepatocellular carcinoma without cirrhosis. Porphyria Cutanea presents with skin fragility and blisters, and patients are at risk of hepatocellular carcinoma with liver iron overload. Erythropoietic protoporphyria and X-linked protoporphyria present with acute painful photosensitivity, and patients are at risk of acute liver failure. Altogether, porphyrias are still underdiagnosed, but once they are suspected, early diagnosis based on measurement of biochemical metabolites that accumulate in the blood, urine, or feces is essential so specific treatment can be started as soon as possible and long-term liver complications are prevented. Screening families to identify presymptomatic carriers is also crucial to prevent overt disease and chronic hepatic complications. |
Homedan, Chadi; Schmitt, Caroline; Laafi, Jihane; Gueguen, Naïg; Desquiret-Dumas, Valérie; Lenglet, Hugo; Karim, Zoubida; Gouya, Laurent; Deybach, Jean-Charles; Simard, Gilles; Puy, Hervé; Malthièry, Yves; Reynier, Pascal Mitochondrial energetic defects in muscle and brain of a Hmbs-/- mouse model of acute intermittent porphyria Article de journal Dans: Human Molecular Genetics, vol. 24, no. 17, p. 5015–5023, 2015, ISSN: 1460-2083. @article{homedan_mitochondrial_2015,Acute intermittent porphyria (AIP), an autosomal dominant metabolic disease (MIM #176000), is due to a deficiency of hydroxymethylbilane synthase (HMBS), which catalyzes the third step of the heme biosynthetic pathway. The clinical expression of the disease is mainly neurological, involving the autonomous, central and peripheral nervous systems. We explored mitochondrial oxidative phosphorylation (OXPHOS) in the brain and skeletal muscle of the Hmbs(-/-) mouse model first in the basal state (BS), and then after induction of the disease with phenobarbital and treatment with heme arginate (HA). The modification of the respiratory parameters, determined in mice in the BS, reflected a spontaneous metabolic energetic adaptation to HMBS deficiency. Phenobarbital induced a sharp alteration of the oxidative metabolism with a significant decrease of ATP production in skeletal muscle that was restored by treatment with HA. This OXPHOS defect was due to deficiencies in complexes I and II in the skeletal muscle whereas all four respiratory chain complexes were affected in the brain. To date, the pathogenesis of AIP has been mainly attributed to the neurotoxicity of aminolevulinic acid and heme deficiency. Our results show that mitochondrial energetic failure also plays an important role in the expression of the disease. |
Pallet, Nicolas; Mami, Iadh; Schmitt, Caroline; Karim, Zoubida; François, Arnaud; Rabant, Marion; Nochy, Dominique; Gouya, Laurent; Deybach, Jean-Charles; Xu-Dubois, Yichum; Thervet, Eric; Puy, Hervé; Karras, Alexandre High prevalence of and potential mechanisms for chronic kidney disease in patients with acute intermittent porphyria Article de journal Dans: Kidney International, vol. 88, no. 2, p. 386–395, 2015, ISSN: 1523-1755. @article{pallet_high_2015,Acute intermittent porphyria (AIP) is a genetic disorder of the synthesis of heme caused by a deficiency in hydroxymethylbilane synthase (HMBS), leading to the overproduction of the porphyrin precursors δ-aminolevulinic acid and porphobilinogen. The aim of this study is to describe the clinical and biological characteristics, the renal pathology, and the cellular mechanisms of chronic kidney disease associated with AIP. A total of 415 patients with HMBS deficiency followed up in the French Porphyria Center were enrolled in 2003 in a population-based study. A follow-up study was conducted in 2013, assessing patients for clinical, biological, and histological parameters. In vitro models were used to determine whether porphyrin precursors promote tubular and endothelial cytotoxicity. Chronic kidney disease occurred in up to 59% of the symptomatic AIP patients, with a decline in the glomerular filtration rate of textasciitilde1 ml/min per 1.73 m(2) annually. Proteinuria was absent in the vast majority of the cases. The renal pathology was a chronic tubulointerstitial nephropathy, associated with a fibrous intimal hyperplasia and focal cortical atrophy. Our experimental data provide evidence that porphyrin precursors promote endoplasmic reticulum stress, apoptosis, and epithelial phenotypic changes in proximal tubular cells. In conclusion, the diagnosis of chronic kidney disease associated with AIP should be considered in cases of chronic tubulointerstitial nephropathy and/or focal cortical atrophy with severe proliferative arteriosclerosis. |
Martelli, Alain; Schmucker, Stéphane; Reutenauer, Laurence; Mathieu, Jacques R. R.; Peyssonnaux, Carole; Karim, Zoubida; Puy, Hervé; Galy, Bruno; Hentze, Matthias W.; Puccio, Hélène Iron regulatory protein 1 sustains mitochondrial iron loading and function in frataxin deficiency Article de journal Dans: Cell Metabolism, vol. 21, no. 2, p. 311–323, 2015, ISSN: 1932-7420. @article{martelli_iron_2015,Mitochondrial iron accumulation is a hallmark of diseases associated with impaired iron-sulfur cluster (Fe-S) biogenesis, such as Friedreich ataxia linked to frataxin (FXN) deficiency. The pathophysiological relevance of the mitochondrial iron loading and the underlying mechanisms are unknown. Using a mouse model of hepatic FXN deficiency in combination with mice deficient for iron regulatory protein 1 (IRP1), a key regulator of cellular iron metabolism, we show that IRP1 activation in conditions of Fe-S deficiency increases the available cytosolic labile iron pool. Surprisingly, our data indicate that IRP1 activation sustains mitochondrial iron supply and function rather than driving detrimental iron overload. Mitochondrial iron accumulation is shown to depend on mitochondrial dysfunction and heme-dependent upregulation of the mitochondrial iron importer mitoferrin-2. Our results uncover an unexpected protective role of IRP1 in pathological conditions associated with altered Fe-S metabolism. |
Bieth, E.; Eddiry, S.; Gaston, V.; Lorenzini, F.; Buffet, A.; Conte Auriol, F.; Molinas, C.; Cailley, D.; Rooryck, C.; Arveiler, B.; Cavaille, J.; Salles, J. P.; Tauber, M. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome Article de journal Dans: Eur J Hum Genet, vol. 23, no. 2, p. 252-5, 2015, ISSN: 1476-5438 (Electronic) 1018-4813 (Linking). @article{RN26, |
Haine, E.; Salles, J. P.; Khau Van Kien, P.; Conte-Auriol, F.; Gennero, I.; Plancke, A.; Julia, S.; Dulac, Y.; Tauber, M.; Edouard, T. Muscle and Bone Impairment in Children With Marfan Syndrome: Correlation With Age and FBN1 Genotype Article de journal Dans: J Bone Miner Res, vol. 30, no. 8, p. 1369-76, 2015, ISSN: 1523-4681 (Electronic) 0884-0431 (Linking). @article{RN22, |
Sales de Gauzy, J.; Gennero, I.; Delrous, O.; Salles, J. P.; Lepage, B.; Accadbled, F. Fasting total ghrelin levels are increased in patients with adolescent idiopathic scoliosis Article de journal Dans: Scoliosis, vol. 10, p. 33, 2015, ISSN: 1748-7161 (Print) 1748-7161 (Linking). @article{RN23, |
Caminade, A. M.; Fruchon, S.; Turrin, C. O.; Poupot, M.; Ouali, A.; Maraval, A.; Garzoni, M.; Maly, M.; Furer, V.; Kovalenko, V.; Majoral, J. P.; Pavan, G. M.; Poupot, R. The key role of the scaffold on the efficiency of dendrimer nanodrugs Article de journal Dans: Nat Commun, vol. 6, p. 7722, 2015, ISSN: 2041-1723 (Electronic) 2041-1723 (Linking). @article{RN29, |
Fruchon, S.; Mouriot, S.; Thiollier, T.; Grandin, C.; Caminade, A. M.; Turrin, C. O.; Contamin, H.; Poupot, R. Repeated intravenous injections in non-human primates demonstrate preclinical safety of an anti-inflammatory phosphorus-based dendrimer Article de journal Dans: Nanotoxicology, vol. 9, no. 4, p. 433-41, 2015, ISSN: 1743-5404 (Electronic) 1743-5390 (Linking). @article{RN31, |
Hameau, Aurélien; Fruchon, Séverine; Bijani, Christian; Barducci, Alessandro; Blanzat, Muriel; Poupot, Rémy; Pavan, Giovanni M.; Caminade, Anne-Marie; Turrin, Cédric-Olivier Dans: Journal of Polymer Science Part A: Polymer Chemistry, vol. 53, no. 6, p. 761-774, 2015, ISSN: 0887624X. @article{RN40, |
Ledall, J.; Fruchon, S.; Garzoni, M.; Pavan, G. M.; Caminade, A. M.; Turrin, C. O.; Blanzat, M.; Poupot, R. Interaction studies reveal specific recognition of an anti-inflammatory polyphosphorhydrazone dendrimer by human monocytes Article de journal Dans: Nanoscale, vol. 7, no. 42, p. 17672-84, 2015, ISSN: 2040-3372 (Electronic) 2040-3364 (Linking). @article{RN35, |
Bruyere, J.; Roy, E.; Ausseil, J.; Lemonnier, T.; Teyre, G.; Bohl, D.; Etienne-Manneville, S.; Lortat-Jacob, H.; Heard, J. M.; Vitry, S. Heparan sulfate saccharides modify focal adhesions: implication in mucopolysaccharidosis neuropathophysiology Article de journal Dans: J Mol Biol, vol. 427, no. 4, p. 775-791, 2015, ISSN: 1089-8638 (Electronic) 0022-2836 (Linking). @article{RN1024, |
Martins, C.; Hulkova, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F. L.; Ohmi, K.; DiCristo, G.; Hamel, E.; Ausseil, J.; Cheillan, D.; Moreau, A.; Svobodova, E.; Hajkova, Z.; Tesarova, M.; Hansikova, H.; Bigger, B. W.; Hrebicek, M.; Pshezhetsky, A. V. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model Article de journal Dans: Brain, vol. 138, no. Pt 2, p. 336-55, 2015, ISSN: 1460-2156 (Electronic) 0006-8950 (Linking). @article{RN1021, |
Trudel, S.; Trecherel, E.; Gomila, C.; Peltier, M.; Aubignat, M.; Gubler, B.; Morliere, P.; Heard, J. M.; Ausseil, J. Oxidative stress is independent of inflammation in the neurodegenerative Sanfilippo syndrome type B Article de journal Dans: J Neurosci Res, vol. 93, no. 3, p. 424-32, 2015, ISSN: 1097-4547 (Electronic) 0360-4012 (Linking). @article{RN1023, |
2014 |
Homedan, Chadi; Laafi, Jihane; Schmitt, Caroline; Gueguen, Naïg; Lefebvre, Thibaud; Karim, Zoubida; Desquiret-Dumas, Valérie; Wetterwald, Céline; Deybach, Jean-Charles; Gouya, Laurent; Puy, Hervé; Reynier, Pascal; Malthièry, Yves Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model Article de journal Dans: The International Journal of Biochemistry & Cell Biology, vol. 51, p. 93–101, 2014, ISSN: 1878-5875. @article{homedan_acute_2014,Acute intermittent porphyria (AIP), an inherited hepatic disorder, is due to a defect of hydroxymethylbilane synthase (HMBS), an enzyme involved in heme biosynthesis. AIP is characterized by recurrent, life-threatening attacks at least partly due to the increased hepatic production of 5-aminolaevulinic acid (ALA). Both the mitochondrial enzyme, ALA synthase (ALAS) 1, involved in the first step of heme biosynthesis, which is closely linked to mitochondrial bioenergetic pathways, and the promise of an ALAS1 siRNA hepatic therapy in humans, led us to investigate hepatic energetic metabolism in Hmbs KO mice treated with phenobarbital. The mitochondrial respiratory chain (RC) and the tricarboxylic acid (TCA) cycle were explored in the Hmbs(-/-) mouse model. RC and TCA cycle were significantly affected in comparison to controls in mice treated with phenobarbital with decreased activities of RC complexes I (-52%, (**)ptextless0.01), II (-50%, (**)ptextless0.01) and III (-55%, (*)ptextless0.05), and decreased activity of α-ketoglutarate dehydrogenase (-64%, (*)ptextless0.05), citrate synthase (-48%, (**)ptextless0.01) and succinate dehydrogenase (-53%, (*)ptextless0.05). Complex II-driven succinate respiration was also significantly affected. Most of these metabolic alterations were at least partially restored after the phenobarbital arrest and heme arginate administration. These results suggest a cataplerosis of the TCA cycle induced by phenobarbital, caused by the massive withdrawal of succinyl-CoA by ALAS induction, such that the TCA cycle is unable to supply the reduced cofactors to the RC. This profound and reversible impact of AIP on mitochondrial energetic metabolism offers new insights into the beneficial effect of heme, glucose and ALAS1 siRNA treatments by limiting the cataplerosis of TCA cycle. |
Oustric, Vincent; Manceau, Hana; Ducamp, Sarah; Soaid, Rima; Karim, Zoubida; Schmitt, Caroline; Mirmiran, Arienne; Peoc'h, Katell; Grandchamp, Bernard; Beaumont, Carole; Lyoumi, Said; Moreau-Gaudry, François; Guyonnet-Dupérat, Véronique; de Verneuil, Hubert; Marie, Joëlle; Puy, Herve; Deybach, Jean-Charles; Gouya, Laurent Antisense oligonucleotide-based therapy in human erythropoietic protoporphyria Article de journal Dans: American Journal of Human Genetics, vol. 94, no. 4, p. 611–617, 2014, ISSN: 1537-6605. @article{oustric_antisense_2014,In 90% of people with erythropoietic protoporphyria (EPP), the disease results from the inheritance of a common hypomorphic FECH allele, encoding ferrochelatase, in trans to a private deleterious FECH mutation. The activity of the resulting FECH enzyme falls below the critical threshold of 35%, leading to the accumulation of free protoporphyrin IX (PPIX) in bone marrow erythroblasts and in red cells. The mechanism of low expression involves a biallelic polymorphism (c.315-48TtextgreaterC) localized in intron 3. The 315-48C allele increases usage of the 3' cryptic splice site between exons 3 and 4, resulting in the transcription of an unstable mRNA with a premature stop codon, reducing the abundance of wild-type FECH mRNA, and finally reducing FECH activity. Through a candidate-sequence approach and an antisense-oligonucleotide-tiling method, we identified a sequence that, when targeted by an antisense oligonucleotide (ASO-V1), prevented usage of the cryptic splice site. In lymphoblastoid cell lines derived from symptomatic EPP subjects, transfection of ASO-V1 reduced the usage of the cryptic splice site and efficiently redirected the splicing of intron 3 toward the physiological acceptor site, thereby increasing the amount of functional FECH mRNA. Moreover, the administration of ASO-V1 into developing human erythroblasts from an overtly EPP subject markedly increased the production of WT FECH mRNA and reduced the accumulation of PPIX to a level similar to that measured in asymptomatic EPP subjects. Thus, EPP is a paradigmatic Mendelian disease in which the in vivo correction of a common single splicing defect would improve the condition of most affected individuals. |
Cadoudal, T.; Buleon, M.; Sengenes, C.; Diene, G.; Desneulin, F.; Molinas, C.; Eddiry, S.; Conte-Auriol, F.; Daviaud, D.; Martin, P. G.; Bouloumie, A.; Salles, J. P.; Tauber, M.; Valet, P. Impairment of adipose tissue in Prader-Willi syndrome rescued by growth hormone treatment Article de journal Dans: Int J Obes (Lond), vol. 38, no. 9, p. 1234-40, 2014, ISSN: 1476-5497 (Electronic) 0307-0565 (Linking). @article{RN28, |
Y, Degboé; S, Fruchon; M, Baron; D, Nigon; CO, Turrin; AM, Caminade; R, Poupot; A, Cantagrel; JL, Davignon Modulation of pro-inflammatory activation of monocytes and dendritic cells by aza-bis-phosphonate dendrimer as an experimental therapeutic agent Article de journal Dans: Arthritis Research and Therapy, vol. 16, no. R98, 2014. @article{RN30, |
2013 |
Delaby, Constance; Oustric, Vincent; Schmitt, Caroline; Muzeau, Francoise; Robreau, Anne-Marie; Letteron, Philippe; Couchi, Eric; Yu, Angel; Lyoumi, Saïd; Deybach, Jean-Charles; Puy, Herve; Karim, Zoubida; Beaumont, Carole; Grandchamp, Bernard; Demant, Peter; Gouya, Laurent Epistasis in iron metabolism: complex interactions between Cp, Mon1a, and Slc40a1 loci and tissue iron in mice Article de journal Dans: Mammalian Genome: Official Journal of the International Mammalian Genome Society, vol. 24, no. 11-12, p. 427–438, 2013, ISSN: 1432-1777. @article{delaby_epistasis_2013,Disorders of iron metabolism are among the most common acquired and constitutive diseases. Hemochromatosis has a solid genetic basis and in Northern European populations it is usually associated with homozygosity for the C282Y mutation in the HFE protein. However, the penetrance of this mutation is incomplete and the clinical presentation is highly variable. The rare and common variants identified so far as genetic modifiers of HFE-related hemochromatosis are unable to account for the phenotypic heterogeneity of this disorder. There are wide variations in the basal iron status of common inbred mouse strains, and this diversity may reflect the genetic background of the phenotypic diversity under pathological conditions. We therefore examined the genetic basis of iron homeostasis using quantitative trait loci mapping applied to the HcB-15 recombinant congenic strains for tissue and serum iron indices. Two highly significant QTL containing either the N374S Mon1a mutation or the Ferroportin locus were found to be major determinants in spleen and liver iron loading. Interestingly, when considering possible epistatic interactions, the effects of Mon1a on macrophage iron export are conditioned by the genotype at the Slc40a1 locus. Only mice that are C57BL/10ScSnA homozygous at both loci display a lower spleen iron burden. Furthermore, the liver-iron lowering effect of the N374S Mon1a mutation is observed only in mice that display a nonsense mutation in the Ceruloplasmin (Cp) gene. This study highlights the existence of genetic interactions between Cp, Mon1a, and the Slc40a1 locus in iron metabolism, suggesting that epistasis may be a crucial determinant of the variable biological and clinical presentations in iron disorders. |
Lyoumi, Said; Lefebvre, Thibaud; Karim, Zoubida; Gouya, Laurent; Puy, Hervé PXR-ALAS1: a key regulatory pathway in liver toxicity induced by isoniazid-rifampicin antituberculosis treatment Article de journal Dans: Clinics and Research in Hepatology and Gastroenterology, vol. 37, no. 5, p. 439–441, 2013, ISSN: 2210-741X. @article{lyoumi_pxr-alas1_2013, |
Moulouel, Boualem; Houamel, Dounia; Delaby, Constance; Tchernitchko, Dimitri; Vaulont, Sophie; Letteron, Philippe; Thibaudeau, Olivier; Puy, Hervé; Gouya, Laurent; Beaumont, Carole; Karim, Zoubida Hepcidin regulates intrarenal iron handling at the distal nephron Article de journal Dans: Kidney International, vol. 84, no. 4, p. 756–766, 2013, ISSN: 1523-1755. @article{moulouel_hepcidin_2013,Hepcidin, the key regulatory hormone of iron homeostasis, and iron carriers such as transferrin receptor1 (TFR1), divalent metal transporter1 (DMT1), and ferroportin (FPN) are expressed in kidney. Whether hepcidin plays an intrinsic role in the regulation of renal iron transport is unknown. Here, we analyzed the renal handling of iron in hemochromatosis Hepc(-/-) and Hjv(-/-) mouse models, as well as in phenylhydrazine (PHZ)-treated mice. We found a marked medullary iron deposition in the kidneys of Hepc(-/-) mice, and iron leak in the urine. The kidneys of Hepc(-/-) mice exhibited a concomitant decrease in TFR1 and increase in ferritin and FPN expression. Increased FPN abundance was restricted to the thick ascending limb (TAL). DMT1 protein remained unaffected despite a significant decrease of its mRNA level, suggesting that DMT1 protein is stabilized in the absence of hepcidin. Treatment of kidney sections from Hepc(-/-) mice with hepcidin decreased DMT1 protein, an effect confirmed in renal cell lines where hepcidin markedly decreased (55)Fe transport. In the kidneys of Hjv(-/-) mice exhibiting low hepcidin expression, the iron overload was similar to that in the kidneys of Hepc(-/-) mice. However, in PHZ mice, iron accumulation resulting from hemoglobin leak was detected in the proximal tubule. Thus, kidneys exhibit a tissue-specific handling of iron that depends on the extra iron source. Hepcidin may control the expression of iron transporters to prevent renal iron overload. |
Salles, J. P.; Laurencin-Dalicieux, S.; Conte-Auriol, F.; Briand-Mesange, F.; Gennero, I. Bone defects in LPA receptor genetically modified mice Article de journal Dans: Biochim Biophys Acta, vol. 1831, no. 1, p. 93-8, 2013, ISSN: 0006-3002 (Print) 0006-3002 (Linking). @article{RN37, |
Davignon, J. L.; Hayder, M.; Baron, M.; Boyer, J. F.; Constantin, A.; Apparailly, F.; Poupot, R.; Cantagrel, A. Targeting monocytes/macrophages in the treatment of rheumatoid arthritis Article de journal Dans: Rheumatology (Oxford), vol. 52, no. 4, p. 590-8, 2013, ISSN: 1462-0332 (Electronic) 1462-0324 (Linking). @article{RN15b, |
Fruchon, S.; Caminade, A. M.; Abadie, C.; Davignon, J. L.; Combette, J. M.; Turrin, C. O.; Poupot, R. An azabisphosphonate-capped poly(phosphorhydrazone) dendrimer for the treatment of endotoxin-induced uveitis Article de journal Dans: Molecules, vol. 18, no. 8, p. 9305-16, 2013, ISSN: 1420-3049 (Electronic) 1420-3049 (Linking). @article{RN20, |
2012 |
De Rocca Serra-Nedelec, A.; Edouard, T.; Treguer, K.; Tajan, M.; Araki, T.; Dance, M.; Mus, M.; Montagner, A.; Tauber, M.; Salles, J. P.; Valet, P.; Neel, B. G.; Raynal, P.; Yart, A. Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature Article de journal Dans: Proc Natl Acad Sci U S A, vol. 109, no. 11, p. 4257-62, 2012, ISSN: 1091-6490 (Electronic) 0027-8424 (Linking). @article{RN16b, |
Boyer, J. F.; Bongard, V.; Cantagrel, A.; Jamard, B.; Gottenberg, J. E.; Mariette, X.; Davignon, J. L.; Ferrieres, J.; Ruidavets, J. B.; Dallongeville, J.; Arveiler, D.; Cambon-Thomsen, A.; Constantin, A. Link between traditional cardiovascular risk factors and inflammation in patients with early arthritis: results from a French multicenter cohort Article de journal Dans: Arthritis Care Res (Hoboken), vol. 64, no. 6, p. 872-80, 2012, ISSN: 2151-4658 (Electronic) 2151-464X (Linking). @article{RN12b, |
2011 |
Lyoumi, Saïd; Abitbol, Marie; Rainteau, Dominique; Karim, Zoubida; Bernex, Florence; Oustric, Vincent; Millot, Sarah; Lettéron, Philippe; Heming, Nicholas; Guillmot, Laurent; Montagutelli, Xavier; Berdeaux, Gilles; Gouya, Laurent; Poupon, Raoul; Deybach, Jean-Charles; Beaumont, Carole; Puy, Hervé Protoporphyrin retention in hepatocytes and Kupffer cells prevents sclerosing cholangitis in erythropoietic protoporphyria mouse model Article de journal Dans: Gastroenterology, vol. 141, no. 4, p. 1509–1519, 1519.e1–3, 2011, ISSN: 1528-0012. @article{lyoumi_protoporphyrin_2011,BACKGROUND & AIMS: Chronic, progressive hepatobiliary disease is the most severe complication of erythropoietic protoporphyria (EPP) and can require liver transplantation, although the mechanisms that lead to liver failure are unknown. We characterized protoporphyrin-IX (PPIX)-linked hepatobiliary disease in BALB/c and C57BL/6 (Fechm1Pas) mice with mutations in ferrochelatase as models for EPP. METHODS: Fechm1Pas and wild-type (control) mice were studied at 12-14 weeks of age. PPIX was quantified; its distribution in the liver, serum levels of lipoprotein-X, liver histology, contents of bile salt and cholesterol phospholipids, and expression of genes were compared in mice of the BALB/c and C57BL/6 backgrounds. The in vitro binding affinity of PPIX for bile components was determined. RESULTS: Compared with mice of the C57BL/6 background, BALB/c Fechm1Pas mice had a more severe pattern of cholestasis, fibrosis with portoportal bridging, bile acid regurgitation, sclerosing cholangitis, and hepatolithiasis. In C57BL/6 Fechm1Pas mice, PPIX was sequestrated mainly in the cytosol of hepatocytes and Kupffer cells, whereas, in BALB/c Fechm1Pas mice, PPIX was localized within enlarged bile canaliculi. Livers of C57BL/6 Fechm1Pas mice were protected through a combination of lower efflux of PPIX and reduced synthesis and export of bile acid. CONCLUSIONS: PPIX binds to bile components and disrupts the physiologic equilibrium of phospholipids, bile acids, and cholesterol in bile. This process might be involved in pathogenesis of sclerosing cholangitis from EPP; a better understanding might improve diagnosis and development of reagents to treat or prevent liver failure in patients with EPP. |
Brasse-Lagnel, Carole; Karim, Zoubida; Letteron, Philippe; Bekri, Soumeya; Bado, André; Beaumont, Carole Intestinal DMT1 cotransporter is down-regulated by hepcidin via proteasome internalization and degradation Article de journal Dans: Gastroenterology, vol. 140, no. 4, p. 1261–1271.e1, 2011, ISSN: 1528-0012. @article{brasse-lagnel_intestinal_2011,BACKGROUNDS & AIMS: The mechanism by which hepcidin regulates iron export from macrophages has been well established and is believed to involve degradation of ferroportin. However, in the small intestine, hepcidin's mechanisms of action are not known. We studied human polarized intestinal (Caco-2/TC7) cells and mouse duodenal segments, ex vivo, to investigate the molecular mechanisms by which hepcidin down-regulates intestinal transepithelial iron transport. METHODS: Iron transport was analyzed using ⁵⁵FeNTA. Expression of Divalent Metal Transporter 1 (DMT1) and ferroportin was evaluated by reverse-transcription quantitative polymerase chain reaction and immunoblotting. Videomicroscopy analysis was performed on live cells that expressed either DMT1 or ferroportin fused to green fluorescent protein. RESULTS: In Caco-2/TC7 cells, physiologic doses of hepcidin (50-1000 nmol/L) inhibited transport of ⁵⁵Fe in a dose-dependent manner; a half-maximum effect was observed at 75-100 nmol/L. However, 200 nmol/L hepcidin induced a significant decrease in DMT1 protein expression but no change in ferroportin protein levels, unlike macrophages. This result was confirmed ex vivo in isolated duodenal segments: 200 nmol/L hepcidin induced a significant reduction in iron transport and DMT1 protein levels but no change in ferroportin levels. In Caco-2/TC7 cells, the effect of hepcidin on the DMT1 protein level was completely abolished in the presence of a proteasome inhibitor (MG-132); DMT1 ubiquitination was induced by the addition of hepcidin. CONCLUSIONS: An acute increase in hepcidin concentration reduces intestinal iron absorption through ubiquitin-dependent proteasome degradation of DMT1. |
Gennero, I.; Laurencin-Dalicieux, S.; Conte-Auriol, F.; Briand-Mesange, F.; Laurencin, D.; Rue, J.; Beton, N.; Malet, N.; Mus, M.; Tokumura, A.; Bourin, P.; Vico, L.; Brunel, G.; Oreffo, R. O.; Chun, J.; Salles, J. P. Absence of the lysophosphatidic acid receptor LPA1 results in abnormal bone development and decreased bone mass Article de journal Dans: Bone, vol. 49, no. 3, p. 395-403, 2011, ISSN: 1873-2763 (Electronic) 1873-2763 (Linking). @article{RN21b, |
Boyer, J. F.; Gourraud, P. A.; Cantagrel, A.; Davignon, J. L.; Constantin, A. Traditional cardiovascular risk factors in rheumatoid arthritis: a meta-analysis Article de journal Dans: Joint Bone Spine, vol. 78, no. 2, p. 179-83, 2011, ISSN: 1778-7254 (Electronic) 1297-319X (Linking). @article{RN27, |
Courties, G.; Baron, M.; Presumey, J.; Escriou, V.; van Lent, P.; Scherman, D.; Cantagrel, A.; van den Berg, W. B.; Jorgensen, C.; Apparailly, F.; Davignon, J. L. Cytosolic phospholipase A2alpha gene silencing in the myeloid lineage alters development of Th1 responses and reduces disease severity in collagen-induced arthritis Article de journal Dans: Arthritis Rheum, vol. 63, no. 3, p. 681-90, 2011, ISSN: 1529-0131 (Electronic) 0004-3591 (Linking). @article{RN13b, |
Hayder, Myriam; Poupot, Mary; Baron, Michel; Nigon, Delphine; Turrin, Cédric-Olivier; Caminade, Anne-Marie; Majoral, Jean-Pierre; Eisenberg, Robert A.; Fournié, Jean-Jacques; Cantagrel, Alain; Poupot, Rémy; Davignon, Jean-Luc A Phosphorus-Based Dendrimer Targets Inflammation and Osteoclastogenesis in Experimental Arthritis Article de journal Dans: Sci. Transl. Med, vol. 3, no. 81 81ra35, 2011. @article{RN34, |
Hayder, Myriam; Poupot, Mary; Baron, Michel; Nigon, Delphine; Turrin, Cédric-Olivier; Caminade, Anne-Marie; Majoral, Jean-Pierre; Eisenberg, Robert A.; Fournié, Jean-Jacques; Cantagrel, Alain; Poupot, Rémy; Davignon, Jean-Luc A Phosphorus-Based Dendrimer Targets Inflammation and Osteoclastogenesis in Experimental Arthritis Article de journal Dans: Sci. Transl. Med, vol. 3, no. 81 81ra35, 2011. @article{RN34b, |
2010 |
Millot, Sarah; Andrieu, Valérie; Letteron, Philippe; Lyoumi, Saïd; Hurtado-Nedelec, Margarita; Karim, Zoubida; Thibaudeau, Olivier; Bennada, Samira; Charrier, Jean-Luc; Lasocki, Sigismond; Beaumont, Carole Erythropoietin stimulates spleen BMP4-dependent stress erythropoiesis and partially corrects anemia in a mouse model of generalized inflammation Article de journal Dans: Blood, vol. 116, no. 26, p. 6072–6081, 2010, ISSN: 1528-0020. @article{millot_erythropoietin_2010,Mouse bone marrow erythropoiesis is homeostatic, whereas after acute anemia, bone morphogenetic protein 4 (BMP4)-dependent stress erythropoiesis develops in the spleen. The aim of this work was to compare spleen stress erythropoiesis and bone marrow erythropoiesis in a mouse model of zymosan-induced generalized inflammation, which induces long-lasting anemia and to evaluate the ability of erythropoietin (Epo) injections to correct anemia in this setting. The effects of zymosan and/or Epo injections on erythroid precursor maturation and apoptosis, serum interferon-γ levels, hematologic parameters, and spleen BMP4 expression were analyzed, as well as the effect of zymosan on red blood cell half-life. We found that bone marrow erythropoiesis is suppressed by inflammation and does not respond to Epo administration, despite repression of erythroblast apoptosis. On the contrary, a robust erythropoietic response takes place in the spleen after Epo injections in both control and zymosan-induced generalized inflammation mice. This specific response implies Epo-mediated induction of BMP4 expression by F4/80(+) spleen macrophages, proliferation of stress burst-forming units-erythroid, and increased number of spleen erythroblasts. It allows only partial recovery of anemia, probably because of peripheral destruction of mature red cells. It is not clear whether similar BMP4-dependent stress erythropoiesis can occur in human bone marrow after Epo injections. |
Davignon, J. L.; Boyer, J. F.; Jamard, B.; Nigon, D.; Constantin, A.; Cantagrel, A. Maintenance of cytomegalovirus-specific CD4pos T-cell response in rheumatoid arthritis patients receiving anti-tumor necrosis factor treatments Article de journal Dans: Arthritis Res Ther, vol. 12, no. 4, p. R142, 2010, ISSN: 1478-6362 (Electronic) 1478-6354 (Linking). @article{RN14b, |
2009 |
Szutkowska, Marta; Vernimmen, Catherine; Debaix, Huguette; Devuyst, Olivier; Friedlander, Gérard; Karim, Zoubida Zeta-crystallin mediates the acid pH-induced increase of BSC1 cotransporter mRNA stability Article de journal Dans: Kidney International, vol. 76, no. 7, p. 730–738, 2009, ISSN: 1523-1755. @article{szutkowska_zeta-crystallin_2009,The Na+/K+/2Cl- cotransporter (BSC1/NKCC2) is the major transporter mediating sodium chloride and ammonium absorption in the medullary thick ascending limb. A loss-of-function mutation of BSC1 is responsible for Bartter's syndrome. We previously showed both in vivo and in vitro that acidosis increases the expression and activity of BSC1 and that acid pH enhances the stability of BSC1 mRNA by mechanisms involving its 3'-untranslated region (UTR). zeta-Crystallin is a pH response factor that protects the mitochondrial glutaminase mRNA by a specific interaction with AU-rich motifs. Here we identified the molecular determinant(s) within the 3'-UTR that are responsible for BSC1-mRNA expression and assessed the involvement of zeta-crystallin in this regulation. Deleting three out of six conserved AU-rich motifs drastically reduced the expression of BSC1-mRNA with maximal effect for motif 3 at position 870 of the 3'UTR. This motif was responsible for pH and zeta-crystallin-induced stability of BSC1 mRNA. The abundance of zeta-crystallin was increased by acid pH and its overexpression increased the stability of BSC1 mRNA, but its RNA silencing inhibited acid pH-induced BSC1 expression. Therefore the 3'UTR of BSC1-mRNA is a target for zeta-crystallin. The induction of zeta-crystallin by an acid pH plays an important role in preventing BSC1 mRNA decay, thus increasing its expression and activity. |
2008 |
Karim, Zoubida; Gérard, Bénédicte; Bakouh, Naziha; Alili, Rohia; Leroy, Christine; Beck, Laurent; Silve, Caroline; Planelles, Gabrielle; Urena-Torres, Pablo; Grandchamp, Bernard; Friedlander, Gérard; Prié, Dominique NHERF1 mutations and responsiveness of renal parathyroid hormone Article de journal Dans: The New England Journal of Medicine, vol. 359, no. 11, p. 1128–1135, 2008, ISSN: 1533-4406. @article{karim_nherf1_2008,Impaired renal phosphate reabsorption, as measured by dividing the tubular maximal reabsorption of phosphate by the glomerular filtration rate (TmP/GFR), increases the risks of nephrolithiasis and bone demineralization. Data from animal models suggest that sodium-hydrogen exchanger regulatory factor 1 (NHERF1) controls renal phosphate transport. We sequenced the NHERF1 gene in 158 patients, 94 of whom had either nephrolithiasis or bone demineralization. We identified three distinct mutations in seven patients with a low TmP/GFR value. No patients with normal TmP/GFR values had mutations. The mutants expressed in cultured renal cells increased the generation of cyclic AMP (cAMP) by parathyroid hormone (PTH) and inhibited phosphate transport. These NHERF1 mutations suggest a previously unrecognized cause of renal phosphate loss in humans. |
2006 |
Gallazzini, Morgan; Karim, Zoubida; Bichara, Maurice Regulation of ROMK (Kir 1.1) channel expression in kidney thick ascending limb by hypertonicity: role of TonEBP and MAPK pathways Article de journal Dans: Nephron. Physiology, vol. 104, no. 4, p. 126–135, 2006, ISSN: 1660-2137. @article{gallazzini_regulation_2006,The present study assessed the mechanisms by which hypertonicity caused by NaCl enhances the renal outer medullary potassium channel (ROMK) mRNA abundance in rat kidney medullary thick ascending limb (MTAL) and in cultured mouse TAL cells. Using the run-off technique, we observed that the ROMK gene transcription rate in nuclei isolated from MTAL fragments was enhanced approximately 40% by a high NaCl medium. In MTAL fragments, hypertonicity (450 mosm) caused by NaCl, not by mannitol or urea, enhanced both ROMK mRNA abundance and tonicity-responsive enhancer binding protein (TonEBP) total abundance and nuclear localization. In an immortalized mouse TAL cell culture in which ROMK is apically expressed, hypertonicity caused by both NaCl and mannitol, not urea, enhanced both ROMK mRNA abundance and TonEBP total abundance and nuclear localization. Confocal microscopy confirmed an increased nuclear translocation of TonEBP in response to NaCl-induced hypertonicity. Finally, inhibition of the p38 MAPK pathway by SB203580 and of the ERK pathway by PD98059 abolished the NaCl-induced stimulation of TonEBP and ROMK. These results establish that mRNA expression of ROMK is augmented in the MTAL by NaCl-induced hypertonicity through stimulation of ROMK gene transcription, and that TonEBP and the p38 MAPK and ERK pathways are involved in this effect. |
Bichara, Maurice; Attmane-Elakeb, Amel; Brown, Dennis; Essig, Marie; Karim, Zoubida; Muffat-Joly, Martine; Micheli, Laetitia; Eude-Le Parco, Isabelle; Cluzeaud, Françoise; Peuchmaur, Michel; Bonvalet, Jean-Pierre; Poirier, Françoise; Farman, Nicolette Exploring the role of galectin 3 in kidney function: a genetic approach Article de journal Dans: Glycobiology, vol. 16, no. 1, p. 36–45, 2006, ISSN: 0959-6658. @article{bichara_exploring_2006,Galectin 3 belongs to a family of glycoconjugate-binding proteins that participate in cellular homeostasis by modulating cell growth, adhesion, and signaling. We studied adult galectin 3 null mutant (Gal 3-/-) and wild-type (WT) mice to gain insights into the role of galectin 3 in the kidney. By immunofluorescence, galectin 3 was found in collecting duct (CD) principal and intercalated cells in some regions of the kidney, as well as in the thick ascending limbs at lower levels. Compared to WT mice, Gal 3-/- mice had approximately 11% fewer glomeruli (p textless 0.04), associated with kidney hypertrophy (p textless 0.006). In clearance experiments, urinary chloride excretion was found to be higher in Gal 3-/- than in WT mice (p textless 0.04), but there was no difference in urinary bicarbonate excretion, in glomerular filtration, or urinary flow rates. Under chronic low sodium diet, Gal 3-/- mice had lower extracellular fluid (ECF) volume than WT mice (p textless 0.05). Plasma aldosterone concentration was higher in Gal 3-/- than in WT mice (p textless 0.04), which probably caused the observed increase in alpha-epithelial sodium channel (alpha-ENaC) protein abundance in the mutant mice (p textless 0.001). Chronic high sodium diet resulted paradoxically in lower blood pressure (p textless 0.01) in Gal 3-/- than in WT. We conclude that Gal 3-/- mice have mild renal chloride loss, which causes chronic ECF volume contraction and reduced blood pressure levels. |
2005 |
Karim, Zoubida; Szutkowska, Marta; Vernimmen, Catherine; Bichara, Maurice Renal handling of NH3/NH4+: recent concepts Article de journal Dans: Nephron. Physiology, vol. 101, no. 4, p. p77–81, 2005, ISSN: 1660-2137. @article{karim_renal_2005,To be appropriately excreted in urine, NH4+, the major component of urinary acid excretion, must be synthesized by proximal tubular cells, secreted into the proximal tubular fluid, reabsorbed by the medullary thick ascending limb (MTAL) to be accumulated in the medullary interstitium, and finally secreted in medullary collecting ducts. Several targets have been identified to account at the gene expression level for the adaptation of renal NH4+ synthesis and transport in response to a chronic acid load. These targets are the key enzymes of ammoniagenesis (mitochondrial glutaminase and glutamate dehydrogenase) and gluconeogenesis (phosphoenolpyruvate carboxykinase) and the Na+/H+(NH4+) exchanger NHE3 in the proximal tubule, the apical Na+-K+(NH4+)-2Cl- cotransporter of the MTAL, the basolateral Na+-K+(NH4+)-2Cl- cotransporter, and likely the epithelial Rh B and C glycoproteins in the collecting ducts. An acid pH per se appears to be a major factor in the control of the expression of these genes during metabolic acidosis probably through activation of pH sensors. Glucocorticoids may also act in concert with an acid pH to coordinate the adaptation of various tubular cell types. The present review focuses on some new aspects of NH3/ NH4+ transport and of regulations of gene expression that have recently emerged. |
2003 |
Karim, Zoubida; Attmane-Elakeb, Amel; Sibella, Valérie; Bichara, Maurice Acid pH increases the stability of BSC1/NKCC2 mRNA in the medullary thick ascending limb Article de journal Dans: Journal of the American Society of Nephrology: JASN, vol. 14, no. 9, p. 2229–2236, 2003, ISSN: 1046-6673. @article{karim_acid_2003,Chronic metabolic acidosis enhances the ability of the medullary thick ascending limb (MTAL) to absorb NH(4)(+) at least in part by stimulating the mRNA and protein expression of BSC1/NKCC2, the MTAL apical Na(+)-K(+)(NH(4)(+))-2Cl(-) co-transporter. For assessing the mechanism by which an acid pH enhances the BSC1 mRNA abundance, MTAL were harvested from adrenalectomized rats and incubated in control (pH 7.35) and acid (pH 7.10) 1:1 mixtures of Ham's nutrient mixture F-12 and DME. rBSC1 mRNA abundance and gene transcription rate were quantified by quantitative reverse transcription-PCR and run-off assay, respectively. Acid incubation enhanced mRNA abundance within 4 h in whole cell (P textless 0.02) but not in nucleus. BSC1 gene transcription rate was not affected by acid incubation. In contrast, under conditions in which gene transcription was blocked, rBSC1 mRNA decreased within 6 h by 38 +/- 11% in control but only by 15 +/- 15% in acid medium (P textless 0.02), which represented an increase in the BSC1 mRNA half-life from approximately 7 to approximately 17 h. Furthermore, in a mouse TAL cell line, acid incubation for 16 h significantly increased (P textless 0.02) the amount of BSC1 mRNA in cells transfected with the full-length mBSC1 cDNA but not in cells transfected with a mBSC1 cDNA lacking the 3'-UTR. These results demonstrate that acid pH enhances the stability of BSC1 mRNA probably by activating pathways that act on the AU-rich 3'-UTR of BSC1 mRNA, which contributes to the renal response to metabolic acidosis. |
2002 |
Karim, Zoubida; Attmane-Elakeb, Amel; Bichara, Maurice Renal handling of NH4+ in relation to the control of acid-base balance by the kidney Article de journal Dans: Journal of Nephrology, vol. 15 Suppl 5, p. S128–134, 2002, ISSN: 1121-8428. @article{karim_renal_2002,The major component of urinary acid excretion is NH4+. To be appropriately excreted in urine, NH4+ must be synthesized by proximal tubular cells, secreted into the proximal tubular fluid, reabsorbed by the medullary thick ascending limb (MTAL) to be accumulated in the medullary interstitium, and finally secreted in medullary collecting ducts. Each step of this renal pathway is highly regulated and, in addition to acute events mediated by peptide hormones such as parathyroid hormone, the control of gene expression explains how the renal handling of NH4+ fully adapts to chronic changes in the acid-base status. Several targets have been identified at the gene expression level to account for the adaptation of renal NH4+ synthesis and transport in response to an acid load. These are the key enzymes of ammoniagenesis (mitochondrial glutaminase and glutamate dehydrogenase) and gluconeogenesis (phosphoenolpyruvate carboxykinase) in the proximal tubule, the apical Na(+)-K+(NH4+)-2Cl- cotransporter of the MTAL, and the basolateral Na(+)-K+(NH4+)-2Cl- cotransporter of medullary collecting ducts. At least two factors control the expression of these genes during metabolic acidosis: an acid pH and glucocorticoids, which appear to act in concert to coordinate the adaptation of various tubular cell types. The present review focuses on some aspects of these regulations that have been recently elucidated. |
Impact sociétal
Les projets de l’équipe sont dédiés à l’étude de la physiopathologie des maladies chroniques inflammatoires et au développement de biothérapies. Une composante importante concerne l’étude de la neuroinflammation dans des maladies neurodégénératives adulte (maladie de Parkinson) ou pédiatrique (syndrome de Sanfilippo) mais également dans des maladies du domaine de l’endocrinologie pédiatrique telles que le syndrome de Prader Willi (PWS). Nous nous intéressons également à l’inflammation de maladies ostéo-articulaires telles que la polyarthrite rhumatoïde. La force de notre équipe est de réunir des chercheurs et des cliniciens du CHU fortement impliqués dans des centres de référence reconnus nationalement et internationalement (centre de référence des maladies métaboliques, centre de référence du PWS, service de rhumatologie du CHU de Toulouse). Cette proximité nous permet d’être moteur dans un certain nombre de protocoles liés à des essais de recherche clinique. Nous sommes également investigateurs principaux de plusieurs essais cliniques en cours de développement (ocytocine dans le PWS, thérapie génique dans le syndrome de Sanfilippo). Enfin, nous développons des approches anti-inflammatoires innovantes utilisant des dendrimères phosphorés qui sont des molécules immuno-modulatrices ciblant les cellules myéloïdes pour une réponse anti-inflammatoire, ou en utilisant des dérivés saccharidiques ciblant le récepteur TLR4 dans le système immunitaire inné. Le ciblage de l’hepcidine et / ou de la récupération du fer est également actuellement étudié en tant que nouvelles approches pour le syndrome de Sanfilippo et la polyarthrite rhumatoïde. Toutes ces molécules offrent des possibilités thérapeutiques non explorées encore pour les maladies inflammatoires chroniques.
Collaborations
Locales :
- Dr. Muriel BLANZAT, Laboratoire des IMRCP, Toulouse.
- Dr. Anne-Marie CAMINADE et Dr. Cédric-Olivier TURRIN, Laboratoire de Chimie de Coordination, Toulouse
- Dr. Nicolas FAZILLEAU, équipe 4, CPTP, Toulouse
- Dr. Camille LAURENT, équipe 7 CRCT, Toulouse
- Pr. Bernard PAYRASTRE, équipe 11, I2MC, Toulouse
- Dr. Véronique PONS, équipe 8, l’I2MC, Toulouse
- Dr. Abdelhadi SAOUDI, équipe 5, CPTP, Toulouse
- Dr. Guillaume TABOURET, École Nationale Vétérinaire de Toulouse, INRA UMR1225, Toulouse
- Dr. Armelle YART, équipe 3, l’I2MC, Toulouse
Nationales :
- Dr. Daniel BOUVARD, Institut Albert Bonniot, Site Santé, GRENOBLE.
- Dr. Hélène CAVÉ, Département de Génétique, CHU Paris – Hôpital Robert Debré, PARIS.
- Fabienne COURY, Département de Rhumatologie, centre hospitalier Lyon Sud, 69495 Pierre-Bénite; Université Lyon 1, 69000
- Lyon, INSERM, UMR1033, SFR Santé Lyon-Est, LYON.
- Dr. Olivier PEYRUCHAUD, INSERM, UMR1033, SFR Santé Lyon-Est, LYON.
Internationales:
- Pr. Jerold CHUN, the Scripps Research institute, Department of Neuroscience, California Campus, La Jolla, USA.
- Pr. Eduardo FERNANDEZ-MEGIA, « Centro Singular de Investigación en Química iolóxica e Materiais Moleculares » (CIQUS), Universidade de Santiago de Compostela, SPAIN.
- Dr. Jane GROGAN, Genentech, 1 DNA Way, South San Francisco, CA 94080, USA.
- Dr. Andrey KRUGLOV, Dr. Sergei NEDOSPASOV, Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, Russia. Lomonosov Moscow State University, Moscow, RUSSIA. German Rheumatism Research Center (DRFZ), Berlin, GERMANY.
- Pr. Rudolph LEIBEL, Division of Molecular Genetics and the Naomi Berrie Diabetes Center, Columbia University, NYC, USA.
- Dr. Giulio G. MUCCIOLI, Université catholique de Louvain, Bruxelles, BELGIUM.
- Dr. Giovanni M. PAVAN, University for Applied Sciences and Arts of Southern Switzerland (SUPSI), Manno, SWITZERLAND.
- Dr. V. Gaëlle ROULLIN, School of Pharmacy, university of Montréal, Québec, CANADA.
- Dr. Timofey ROZHDESTVENSKY, Institute of Experimental Pathology (ZMBE), University of Muenster, Von-Esmarch-Str. 56, D-48149 Münster, GERMANY.